




Pulmonary embolism (PE) affects 0.5–1 per 1000 people in the general population each year and is one of the commonest preventable causes of death among hospital inpatients.1,2 Although anticoagulant therapy is highly effective at preventing death, it is frequently either not administered, or administered too late, because the diagnosis of PE has not been entertained.
This update on venous thromboembolism (VTE) reviews the pathophysiology, diagnosis, prognosis and treatment of PE, and unresolved issues in the management of VTE. The risk factors and primary prevention of VTE were the subject of an earlier article.3
Most episodes of PE result from embolisation of thrombi in the leg or pelvic veins. Very large emboli may lodge at the bifurcation of the pulmonary arteries (“saddle embolus”), leading to rapid circulatory failure or sudden death. However, most lodge in lower-order pulmonary vessels.4
The pathophysiological consequences of PE and corresponding clinical features are presented in Box 1.
Because the lung has no pain fibres, PE only causes chest pain if there is involvement of parietal pleura. Fever is common.
A common approach to patients with cardiopulmonary symptoms is the following thought sequence:
Dyspnoea and/or chest pain and/or cough and/or haemoptysis can occur in PE.
PE can be fatal.
If there is any doubt, it is negligent not to investigate for PE.
This fear of missing PE may account for clinicians being better at excluding PE (incorrect in only 10% of cases) than in diagnosing it (correct in 30% of cases) on clinical grounds.5
The clinical diagnosis of PE is unreliable, because the clinical features (Boxes 1 and 2) are poorly sensitive and poorly specific for the diagnosis.6-10 False-negative diagnoses may arise because the symptoms of PE may mimic those of other common cardiopulmonary conditions (Box 2), or they may be attributed to other coexisting cardiopulmonary diseases, such as congestive cardiac failure and pneumonia.
Because there are no diagnostic clinical features of PE, the role of the clinical assessment is to formulate the patient’s presenting symptoms and signs into an estimate of the pretest probability of PE. This serves to define the strategy for special investigations (Box 3). Experienced clinicians can use clinical judgement (“gestalt”) to assign a clinical pretest probability for the diagnosis of PE with reasonable accuracy. Structured clinical prediction rules perform equally well, and have the advantage that they can be used by less experienced clinicians.5 The Simplified Wells Scoring System is the most widely used, because it is simple and rapid, and classifies patients into low, intermediate, or high risk for PE (Box 4).11
Laboratory tests alone cannot reliably confirm or exclude PE, but they may be useful in conjunction with the clinical pretest probability to exclude the diagnosis, to establish an alternative diagnosis, or for risk stratification.
The role of D-dimer testing in patients with suspected PE is to exclude the diagnosis; normal concentrations of D-dimer have a high negative predictive value for PE, particularly in patients with a low clinical pretest probability.12
The main role of the electrocardiogram (ECG) and chest x-ray (CXR) in patients with suspected PE is to exclude alternative diagnoses (eg, myocardial infarction, pneumothorax).13,14 Classical ECG changes for PE are an S wave in lead I, Q wave and T wave inversion in lead III, and T wave inversion in leads V1 to V4. More commonly, the ECG demonstrates minor non-specific ST segment or T wave changes. The CXR may be normal or show enlarged pulmonary arteries, atelectasis, vascular oligaemia, or, rarely, a wedge-shaped density indicating pulmonary infarction. It is essential to have a good quality postero-anterior erect CXR, and to inspect it rigorously.
Arterial blood gas measurements are still widely used in the initial laboratory evaluation of suspected PE, but are of very little value to establish or exclude the diagnosis. They are normal in 20% of patients with proven PE.
If a patient with suspected PE has concurrent, clinically apparent deep venous thrombosis (DVT), leg ultrasound is usually diagnostic, rendering lung imaging unnecessary to establish the diagnosis of PE. Otherwise, specific lung imaging studies are required to diagnose PE.
Contrast pulmonary angiography is the traditional reference standard test for the diagnosis of pulmonary embolism, but is rarely used: it is invasive, requires a high level of expertise, and is not widely available.4,13
Ventilation perfusion (V/Q) isotope scanning reliably establishes the diagnosis of PE if the V/Q features suggest a high probability of PE, and excludes the diagnosis if the scan is normal. However, more than half the patients with suspected PE have a non-diagnostic scan (low or intermediate probability), and a quarter of these have PE. These indeterminate patients require further evaluation with serial non-invasive imaging of leg veins using compression ultrasound, computed tomography pulmonary angiography (CTPA), magnetic resonance angiography, or conventional pulmonary angiography.13,14 Improvements in the methods of ventilation perfusion scanning using tomographic imaging (SPECT) may overcome the problem of non-diagnostic scans in the future.
The advantages of CTPA are that it is rapid, convenient, non-diagnostic in less than 10% of cases, and can be used to establish alternative diagnoses.13 First-generation CT scanners may fail to detect as many as one-third of pulmonary emboli because they have only 5 mm resolution. However, most emboli that are missed are subsegmental and of uncertain clinical significance. Newer multi-slice scanners provide 1 mm resolution and are likely to be more sensitive for subsegmental emboli.15 The major disadvantage of CTPA is the high radiation dose.
Contrast-enhanced magnetic resonance angiography may replace multi-slice CTPA because it does not involve ionising radiation and the contrast agents are safer.
Our approach to the diagnosis of PE is outlined in Box 3. This enables a definitive diagnosis in most patients with suspected PE.
Patients with a low clinical pretest probability of PE (probability 3.6% based on a score of < 2 in Box 4) should have a D-dimer assay performed in the first instance. If the D-dimer result is negative, diagnostic imaging is not required and it is safe to withhold treatment, as the 3-month cumulative incidence of subsequent venous thromboembolism in untreated patients is very low (0.5%). If the D-dimer level is raised, then the algorithm for a moderate or high pretest probability should be followed.16,17
Patients with a moderate clinical pretest probability of PE (probability 20.5%, based on a score of 2–6 in Box 4) or high clinical pretest probability (probability 67%, based on a score of > 6 in Box 4) should proceed straight to V/Q scan or CTPA, because a negative D-dimer result cannot reliably exclude the diagnosis. Even with a moderate clinical probability, a negative D-dimer result may be associated with an unacceptably high (3.4%) 3-month incidence of venous thromboembolism.11,16,17
A normal V/Q scan excludes the diagnosis and a high probability V/Q scan or positive CTPA confirms the diagnosis of PE. A low or intermediate probability V/Q scan or a normal or subsegmental defect on a first-generation CTPA are non-diagnostic, and should be followed by compression ultrasound of the legs, because these patients have an unacceptably high risk of PE. The presence of DVT on ultrasonography confirms the diagnosis of PE; patients with a negative ultrasound require repeat ultrasonography in 1 week.
Repeat diagnostic imaging at the completion of anticoagulant therapy serves as a baseline reference for patients who may require evaluation for suspected recurrent VTE in the future. Residual thrombus persists in about 50% of patients for many years, and may be misdiagnosed as recurrent VTE in patients who do not have a baseline scan for comparison.
Ten per cent of patients with symptomatic PE die within 1 hour of onset of symptoms.18,19 Among patients who are diagnosed with PE, the mortality rate is about 10% at 2 weeks and 25% at 1 year. However, only 20% of deaths during the first year after PE are a direct consequence of PE; most are due to malignancy and underlying cardiorespiratory disease.18,19
The most important determinants of the clinical outcome of PE are the presence or absence and severity of haemodynamic compromise, a reduction in cardiac output, increase in pulmonary vascular resistance, and right ventricular dysfunction. These are determined, in turn, by the size and location of the emboli, the presence of coexisting cardiopulmonary disease, and the extent of neurohumoral activation in response to the embolus.8-10
Five to 10 per cent of patients with acute PE are haemodynamically unstable at presentation.18 These patients have a mortality rate of 25%, compared with 4% for patients who are haemodynamically stable.8 Half of the haemodynamically stable patients have echocardiographic evidence of right ventricular dysfunction, which is associated with a 15% risk of death in hospital and 8% risk of death at 1 year. This compares with no risk of death in patients who do not have echocardiographic evidence of right ventricular dysfunction at presentation.7,8
Chronic pulmonary hypertension occurs in 4% of patients who are treated for PE.20
The aim of treatment for PE is to relieve symptoms, prevent death, reduce the risk of developing chronic pulmonary hypertension, and prevent recurrence. The evidence supporting the treatment recommendations has been graded according to the National Health and Medical Research Council’s levels of evidence (Box 5).21
Anticoagulation is the mainstay of treatment for PE (E31). The role of pharmacological or mechanical embolectomy is restricted to patients who are haemodynamically unstable at presentation.22
The initial anticoagulant treatment for PE is the same as for DVT: either low-molecular-weight heparin (LMWH) or unfractionated heparin (UFH) (E1).22-24 The risk of major bleeding is 1%–5%, and increases with age greater than 70 years and increasing heparin dose.25 The advantages of LMWH over UFH and contraindications to anticoagulation are detailed in our earlier article.3
In the absence of contraindications, all patients with suspected PE and a high pretest probability should be treated with LMWH or UFH until the diagnosis is confirmed or excluded (E4).
Thrombolysis should not be used in unselected patients with PE, because it causes major bleeding in 10% and intracranial bleeding in 3% without reducing overall mortality (E1).26 In patients who are haemodynamically unstable at presentation, mortality may be reduced and the balance between risk and benefit appears to favour thrombolysis (E2).26,27
In patients with PE who are haemodynamically unstable or shocked and have contraindications or do not respond to thrombolysis, percutaneous transcatheter or surgical embolectomy may be life-saving (E32).28 However, a high level of expertise is required to perform these procedures.
Warfarin is the anticoagulant of choice for the long-term treatment of most patients with VTE, including PE. It is highly effective, reducing the risk of recurrence by 80%–90% (E1).29-31
The target international normalised ratio (INR) for the treatment of VTE is 2.0–3.0 (E2). Higher intensity warfarin treatment (target INR, 3.1–4.0) is not superior to standard intensity warfarin (target INR, 2.0–3.0) for preventing recurrence in high-risk patients, including those with antiphospholipid syndrome (E2).32,33
Warfarin causes major bleeding in 0.5–2.5 per 100 and intracranial or fatal bleeding in 0.1–0.2 per 100 patients per year (E1).29,30 The risk of bleeding increases with increasing age and INR, comorbid conditions, concomitant antiplatelet therapy, and genetic factors such as polymorphisms of the cytochrome P450 system. Although the risk of bleeding is greatest within the first 3 months of warfarin treatment, it is cumulative during long-term treatment.29,30 As a result, there is uncertainty about the optimal duration of warfarin therapy for VTE (E1). In general, treatment should be continued as long as the estimated benefits outweigh the likely risk of bleeding and inconvenience of treatment. The risks and benefits of anticoagulation should be reassessed at least annually.
Patients with provoked VTE have the lowest risk of recurrence; patients with a first episode of unprovoked VTE or a major predisposition have an intermediate risk; and those with a first event plus a major predisposition, recurrent unprovoked events or active malignancy have the highest risk. Among patients with a low or intermediate risk of recurrent VTE (< 10% per year), 3–6 months of anticoagulation therapy is commonly recommended (E1). In those with a high risk of recurrent VTE (> 10% per year), longer term or indefinite anticoagulation therapy is recommended. In patients with cancer, LMWH compared with warfarin reduces the risk of recurrent VTE (E2), but has not been evaluated beyond 6 months.34 In patients at high risk of bleeding, the duration of treatment may need to be shortened.
The rationale for considering a reduced intensity of warfarin for long-term treatment of VTE is to improve the benefit–risk ratio by reducing the risk of bleeding. Although reduced intensity warfarin (target INR, 1.5–2.0) is more effective than placebo in preventing recurrent VTE (E2),35 it is probably less effective than standard intensity warfarin (target INR, 2.0–3.0) and does not cause less bleeding (E2).36 Reducing the intensity of warfarin does not remove the need for laboratory monitoring. Therefore, reduced intensity warfarin is not recommended for long-term prevention of VTE, irrespective of a patient’s baseline risk of recurrence or bleeding (E2).
Important unresolved issues in the management of VTE are summarised in Box 6.
Inherited thrombophilic disorders are independently associated with an increased risk of first-ever and, to a lesser extent, recurrent VTE.37,38 Therefore, it has become routine practice in some centres to perform laboratory testing for inherited thrombophilia in patients who present with a first episode of VTE, particularly when another cause for thrombosis cannot be identified. However, laboratory testing for thrombophilia does not influence the choice, intensity, or duration of anticoagulation therapy in most patients, has not been shown to improve outcomes, and is expensive (costs several hundred dollars, depending on the number of tests that are performed). Screening of family members of affected individuals is also of unproven benefit.
The role of thrombolysis to improve clinical outcomes in haemodynamically stable patients who present with right ventricular dysfunction is unproven. It reduces the risk of clinical deterioration requiring escalation of treatment, but a reduction in death has not been demonstrated (E2).26
The optimal duration of warfarin in patients with a first episode of unprovoked VTE remains controversial, with recommendations of international guidelines ranging from 6 months to indefinite treatment.29
New anticoagulant drugs have been developed to overcome some of the limitations of existing anticoagulants.37,39
Fondaparinux (Arixtra, GlaxoSmithKline) is a synthetic pentasaccharide that is administered by once-daily subcutaneous injection. It does not cause heparin-induced thrombocytopenia, because it does not bind to platelets or platelet factor 4. Furthermore, it has a predictable anticoagulant effect, so laboratory monitoring is not required in most patients. Because fondaparinux is excreted renally, dose adjustment and/or laboratory monitoring is likely to be required in patients with renal impairment. Large randomised trials have demonstrated that fondaparinux is as effective and safe as LMWH for the initial treatment of VTE. Fondaparinux has recently been approved in Australia for the treatment of VTE, but is substantially more expensive than LMWH and is not available on the Pharmaceutical Benefits Scheme.
Ximelagatran (Exanta, AstraZeneca) is an oral direct thrombin inhibitor that is administered twice daily. It is metabolised to the active metabolite melagatran, of which 80% is excreted unchanged by the kidneys.39 Unlike warfarin, ximelagatran has a low potential for drug interactions, and does not require monitoring of anticoagulant effect or dose adjustment in patients with normal renal function. Large randomised trials in patients with VTE have demonstrated a similar incidence of recurrent thrombosis and bleeding in patients treated with ximelagatran compared with enoxaparin for 5 days followed by warfarin, and a significant reduction in recurrent thrombosis in patients treated with 18 months of ximelagatran (after an initial 6 months of warfarin) compared with placebo.39,40 However, the US Food and Drug Administration recently declined to approve ximelagatran for clinical use because of concerns about a 6%–12% incidence of hepatic side effects, which may have caused, or contributed to, several deaths.
A large number of oral direct thrombin inhibitors and direct factor Xa inhibitors are in various stages of clinical development.
1 Pathophysiological consequences and corresponding clinical features of pulmonary embolism
Pathophysiological consequences |
Clinical features (% of patients with feature) |
||||||||||||||
Haemodynamic |
|
||||||||||||||
Reduced cardiac output |
Palpitations (20%) Tachycardia (40%) Hypotension (5%)* |
||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
Increased pulmonary vascular resistance |
|||||||||||||||
Vascular obstruction |
Pulmonary hypertension — loud P2 (25%) Right ventricular dilatation and hypokinesis (50%) Tricuspid regurgitation; neck vein distension (10%) Right ventricular failure (30%) |
||||||||||||||
Neurohumoral agents |
|||||||||||||||
Pulmonary artery baroreceptors |
|||||||||||||||
|
|||||||||||||||
Respiratory |
|
||||||||||||||
Alveolar hyperventilation |
Fever (15%) Cough (10%) Chest pain (60%): pleuritic (40%), substernal (15%) Tachypnoea (60%) Hypocapnia (80%); dyspnoea (60%) Respiratory alkalosis (90%) Raised hemidiaphragm (30%); rales (20%) Consolidation; pleural effusion (40%) Haemoptysis (10%) Atelectasis (30%) |
||||||||||||||
Reflex stimulation of irritant receptors |
|||||||||||||||
Increased airways resistance |
|||||||||||||||
Bronchoconstriction |
|||||||||||||||
Reduced pulmonary compliance |
|||||||||||||||
Lung oedema |
|||||||||||||||
Lung haemorrhage |
|||||||||||||||
Loss of surfactant |
|||||||||||||||
Ventilation–perfusion mismatch causing suboptimal gas exchange |
|||||||||||||||
Increased alveolar dead space |
Hypoxaemia: chest pain/loss of consciousness (80%) |
||||||||||||||
Right-to-left shunting |
Cyanosis (15%) |
||||||||||||||
* Major pulmonary embolism may cause circulatory failure and cardiac arrest. |
|||||||||||||||
2 Differential diagnosis of pulmonary embolism and other frequent causes of dyspnoea, haemoptysis and chest pain6
|
Chest wall |
Pleura |
Parenchyma |
Airways |
Heart |
||||||||||
Dyspnoea |
If saturation on air > 98%, consider hyperventilation |
Often if large effusion or pneumothorax (obvious on CXR) |
If febrile symptoms, CXR changes will be due to pneumonia |
COPD and asthma should be obvious; peak flow will be low |
LVF should be obvious clinically and on CXR |
||||||||||
Haemoptysis |
na |
na |
Due to haemorrhage in PE; CXR usually abnormal |
Acute bronchitis is common; lung cancer less common |
na |
||||||||||
Chest pain |
If tenderness where pain, musculoskeletal cause |
Viral pleurisy is common, often with shoulder pain and/or CXR pleural reaction |
Only if acute disease (visible on CXR) spreading across parietal pleura |
na |
Unless massive PE, central pain may be cardiac ischaemia or pericarditis |
||||||||||
COPD = chronic obstructive pulmonary disease; CXR = chest x-ray; LVF = left ventricular failure; na = not applicable; PE = pulmonary embolism. Conditions commonly (bold, italic) and occasionally (bold) misdiagnosed as acute PE. If respiratory rate < 20/min and normal saturation on air and normal CXR, PE is very unlikely. |
|||||||||||||||
3 Clinical approach to the diagnosis of pulmonary embolism (PE)*
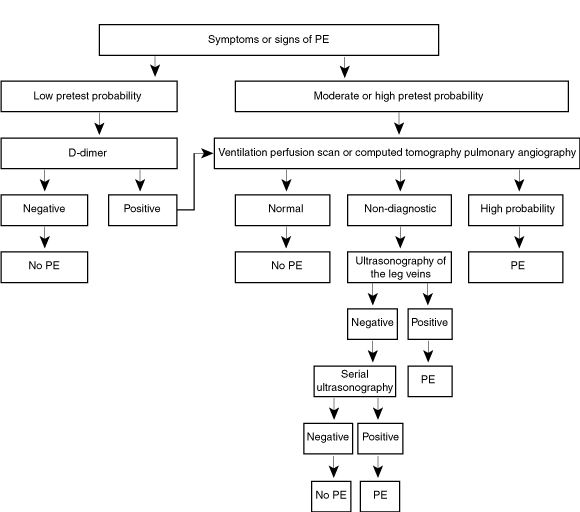
* A subsegmental defect on ventilation perfusion scanning or a normal first generation helical CT scan should be treated as non-diagnostic.
4 Estimating the pretest probability of pulmonary embolism11
Feature |
Score |
||||||||||||||
Clinical signs and symptoms of DVT (minimum of leg swelling and pain with palpation of the deep veins) |
3 points |
||||||||||||||
Alternative diagnosis less likely than PE |
3 points |
||||||||||||||
Heart rate above 100 beats/min |
1.5 points |
||||||||||||||
Immobilisation or surgery in previous 4 weeks |
1.5 points |
||||||||||||||
Previous DVT or PE |
1.5 points |
||||||||||||||
Haemoptysis |
1 point |
||||||||||||||
Cancer |
1 point |
||||||||||||||
DVT = deep venous thrombosis. PE = pulmonary embolism. Low pretest probability if score is < 2; moderate if 2–6 points; high if > 6. |
|||||||||||||||
5 Level-of-evidence codes21
Evidence for the statements made in this article is graded according to the National Health and Medical Research Council (NHMRC) system for assessing the level of evidence.
E1 Level I: Evidence obtained from a systematic review of all relevant randomised controlled trials.
E2 Level II: Evidence obtained from at least one properly designed randomised controlled trial.
E31 Level III-1: Evidence obtained from well-designed pseudo-randomised controlled trials (alternate allocation or some other method).
E32 Level III-2: Evidence obtained from comparative studies with concurrent controls and allocation not randomised (cohort studies), case–control studies, or interrupted time series with a parallel control group.
E33 Level III-3: Evidence obtained from comparative studies with historical control, two or more single-arm studies, or interrupted time series without a parallel control group.
E4 Level IV: Evidence obtained from case-series, either post-test or pretest and post-test.
6 Major unresolved issues in the management of venous thromboembolism
Issue |
Potential benefits |
||||||||||||||
Thrombophilia testing |
Improve risk stratification |
||||||||||||||
Thrombolysis for pulmonary embolism patients with right ventricular dysfunction |
Improve clinical outcome in high-risk patients with pulmonary embolism |
||||||||||||||
Duration of anticoagulants for patients with unprovoked venous thromboembolism |
Optimise patient selection to maximise benefit–risk ratio of extended duration anticoagulation |
||||||||||||||
New anticoagulants (eg, fondaparinux, ximelagatran) |
Overcome the limitations of heparins and warfarin |
||||||||||||||
- 1. Fedullo PF. Pulmonary thromboembolism. In: Murray JF, Nadel JA, Mason RJ, Boushey HA, editors. Textbook of respiratory medicine. 3rd ed. Philadelphia: WB Saunders Company, 2000: 1503-1531.
- 2. Silverstein MD, Heit JA, Mohr DN, et al. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med 1998; 158: 585-593.
- 3. Ho WK, Hankey GJ, Lee CH, Eikelboom JW. Venous thromboembolism: diagnosis and management of deep venous thrombosis. Med J Aust 2005; 182: 476-481. <MJA full text>
- 4. Goldhaber S. Pulmonary embolism. Lancet 2004; 363: 1295-1305.
- 5. Chunilal SD, Eikelboom JW, Attia J, et al. Does this patient have pulmonary embolism? JAMA 2003; 290: 2849-2858.
- 6. Millar AC. Suspected pulmonary embolism. Clin Med 2004; 4: 215-219.
- 7. Dalen JE. Pulmonary embolism: what have we learned since Virchow? Chest 2002; 122: 1440-1456.
- 8. Wood K. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of haemodynamically significant pulmonary embolism. Chest 2002; 121: 877-905.
- 9. Grifoni S, Olivotto I, Cecchini P, et al. Short-term clinical outcome of patients with acute pulmonary embolism, normal blood pressure, and echocardiographic right ventricular dysfunction. Circulation 2000; 101: 2817-2822.
- 10. Goldhaber S, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet 1999; 353: 1386-1389.
- 11. Wells PS, Anderson DR, Rodger M, et al. Excluding pulmonary embolism at the bedside without diagnostic imaging: management of patients with suspected pulmonary embolism presenting to the emergency department by using a simple clinical model and D-dimer. Ann Intern Med 2001; 135: 98-107.
- 12. Kelly J, Hunt BJ. A clinical probability assessment and D-dimer measurement should be the initial step in the investigation of suspected venous thromboembolism. Chest 2003; 124: 1116-1119.
- 13. Pistolesi M, Miniati M. Imaging techniques in treatment algorithms of pulmonary embolism. Eur Respir J 2002; 19 (Suppl 35): 28S-39S.
- 14. Riedel M. Diagnosing pulmonary embolism. Postgrad Med J 2004; 80: 309-319.
- 15. Qanadli SD, Hajjam ME, Mesurolle B, et al. Pulmonary embolism detection: prospective evaluation of dual-section helical CT versus selective pulmonary arteriography in 157 patients. Radiology 2000; 217: 447-455.
- 16. Goldhaber S, Elliot G. Acute pulmonary embolism: part 1. Epidemiology, pathophysiology and diagnosis. Circulation 2003; 108: 2726-2729.
- 17. Stein PD, Hull RD, Patel KC, et al. D-dimer for the exclusion of acute venous thrombosis and pulmonary embolism: a systematic review. Ann Intern Med 2004; 140: 589-602.
- 18. Kearon C. Natural history of venous thromboembolism. Circulation 2003; 107 (Suppl 1): I22-I30.
- 19. Ribeiro A, Lindmarker P, Johnsson H, et al. Pulmonary embolism — one year follow-up with echocardiography doppler and five-year survival analysis. Circulation 1999; 99: 1325-1330.
- 20. Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004; 350: 2257-2264.
- 21. National Health and Medical Research Council. A guide to the development, implementation, and evaluation of clinical practice guidelines. Canberra: NHMRC, 1999.
- 22. McRae S, Ginsberg J. Initial treatment of venous thromboembolism. Circulation 2004; 110 (Suppl 1): I3-I9.
- 23. Quinlan DJ, McQuillan A, Eikelboom JW. Low-molecular-weight heparin compared with intravenous unfractionated heparin for the treatment of pulmonary embolism: a meta-analysis of randomized controlled trials. Ann Intern Med 2004; 140; 175-183.
- 24. Hirsh J, Raschke R. Heparin and low-molecular-weight heparin. The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126: 188S-203S.
- 25. Levine MN, Raskob G, Beyth RJ, et al. Hemorrhagic complications of anticoagulant treatment. The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126: 287S-310S.
- 26. Wan S, Quinlan DJ, Agnelli G, Eikelboom JW. Thrombolysis compared with heparin for initial treatment of pulmonary embolism: a meta-analysis of randomized controlled trials. Circulation 2004; 110: 744-749.
- 27. Dalen JE, Alpert JS, Hirsh J. Thrombolytic therapy for pulmonary embolism: is it effective? Is it safe? When is it indicated? Arch Intern Med 1997; 157: 2550-2556.
- 28. Augustinos P, Ouriel K. Invasive approaches to treatment of venous thromboembolism. Circulation 2004; 110 (Suppl 1): I27-I34.
- 29. Kearon C. Long term management of patients after venous thromboembolism. Circulation 2004; 110 (Suppl 1): I10-I18.
- 30. Buller HR, Agnelli G, Hull RD, et al. Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 (3 Suppl): 401S-428S.
- 31. Goldhaber S, Elliot C. Acute pulmonary embolism: part II: risk stratification, treatment, and prevention. Circulation 2003; 108: 2834-2838.
- 32. Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for prevention of recurrent thrombosis in patients with antiphospholipid antibody syndrome. N Engl J Med 2003; 349: 1133-1138.
- 33. Greaves M, Cohen H, Machin SJ, Mackie I. Guidelines on the investigation and management of the antiphospholipid syndrome. Br J Haematol 2000; 109: 704-715.
- 34. Lee AY, Levine MN, Baker RI, et al; Randomized Comparison of Low-Molecular-Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer (CLOT) Investigators. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349: 146-153.
- 35. Ridker PM, Goldhaber SZ, Danielson E, et al. Long-term, low intensity warfarin therapy for prevention of recurrent venous thromboembolism. N Engl J Med 2003; 348: 1425-1434.
- 36. Kearon C, Ginsberg JS, Kovacs MJ, et al. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349: 631-639.
- 37. Weitz JI, Middeldorp S, Heit JA. Thrombophilia and new anticoagulant drugs. Haematology (Am Soc Hematol Educ Program) 2004; 424-438.
- 38. Lopez JA, Kearon C, Lee AYY. Deep vein thrombosis. Haematology (Am Soc Hematol Educ Program) 2004; 439-456.
- 39. Weitz JI. New anticoagulants for treatment of venous thromboembolism. Circulation 2004; 110 (Suppl 1): I19-I26.
- 40. Brighton T. The direct thrombin inhibitor melagatran/ximelagatran. Med J Aust 2004; 181: 432-437. <MJA full text>
Abstract
Pulmonary embolism (PE) affects 0.5–1 per 1000 people in the general population each year, and is one of the most common preventable causes of death among hospitalised patients.
The clinical diagnosis of PE is unreliable and must be confirmed objectively with ventilation perfusion scanning or computed tomography pulmonary angiography.
The diagnosis of PE can be reliably excluded, without the need for diagnostic imaging, if the clinical pretest probability for PE is low and the D-dimer assay result is negative.
The initial treatment of PE is low-molecular-weight heparin or unfractionated heparin for at least 5 days, followed by warfarin (target international normalised ratio [INR], 2.0–3.0) for at least 3–6 months. Patients with a high clinical pretest probability of PE should commence treatment immediately while awaiting the results of the diagnostic work-up.
Thrombolysis is indicated for patients with objectively confirmed PE who are haemodynamically unstable.
Percutaneous transcatheter or surgical embolectomy may be life-saving in patients ineligible for, or unresponsive to, thrombolytic therapy.
Unresolved issues in the management of venous thromboembolism include the roles of thrombophilia testing, thrombolysis for the treatment of stable PE patients who present with right ventricular dysfunction, and new anticoagulants; and the duration of anticoagulation for first unprovoked venous thromboembolism.