





Volume 208 - Issue 2
Diagnosis and management of idiopathic pulmonary fibrosis: Thoracic Society of Australia and New Zealand and Lung Foundation Australia position statements summary
Authors: Helen E Jo*, Jyotika D Prasad*, Lauren K Troy, Annabelle Mahar, Jane Bleasel, Samantha J Ellis, Daniel C Chambers, Anne E Holland, Fiona R Lake, Gregory Keir, Nicole S Goh, Margaret Wilsher, Sally de Boer, Yuben Moodley, Christopher Grainge, Helen M Whitford, Sally A Chapman, Paul N Reynolds, David Beatson**, Leonie J Jones, Peter Hopkins, Heather M Allan, Ian Glaspole*** and Tamera J Corte***
Med J Aust 2018; 208 (2): 82-88. || doi: 10.5694/mja17.00799
Published online: 20 November 2017
Published online: 20 November 2017
Idiopathic pulmonary fibrosis (IPF) is a fibrosing interstitial lung disease associated with debilitating symptoms of dyspnoea and cough
Abstract
Introduction: Idiopathic pulmonary fibrosis (IPF) is a fibrosing interstitial lung disease associated with debilitating symptoms of dyspnoea and cough, resulting in respiratory failure, impaired quality of life and ultimately death. Diagnosing IPF can be challenging, as it often shares many features with other interstitial lung diseases. In this article, we summarise recent joint position statements on the diagnosis and management of IPF from the Thoracic Society of Australia and New Zealand and Lung Foundation Australia, specifically tailored for physicians across Australia and New Zealand.
Main suggestions:
- A comprehensive multidisciplinary team meeting is suggested to establish a prompt and precise IPF diagnosis.
- Antifibrotic therapies should be considered to slow disease progression. However, enthusiasm should be tempered by the lack of evidence in many IPF subgroups, particularly the broader disease severity spectrum.
- Non-pharmacological interventions including pulmonary rehabilitation, supplemental oxygen, appropriate treatment of comorbidities and disease-related symptoms remain crucial to optimal management.
- Despite recent advances, IPF remains a fatal disease and suitable patients should be referred for lung transplantation assessment.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and idiopathic interstitial lung disease (ILD) which, while demonstrating a heterogeneous clinical course, carries a universally poor prognosis that is worse than many forms of malignancy.1 IPF is more common in older men, and most patients have a history of cigarette smoking.2 The mean survival from diagnosis is only 2–5 years.3
Patients usually experience progressive worsening of dyspnoea on exertion, often with a debilitating dry cough, resulting in significant limitation in their ability to perform activities of daily living and reduction in their quality of life.1
Accurately diagnosing IPF can be challenging, as the pathognomonic, radiological and histopathological findings of usual interstitial pneumonia seen in patients with IPF may also be present in other ILDs, such as connective tissue disease-related ILD, chronic hypersensitivity pneumonitis, asbestosis and drug toxicity.4-6 In 2011, the American Thoracic Society and European Respiratory Society published guidelines for the diagnosis of IPF, recommending that, given these difficulties, IPF be diagnosed within the context of an expert ILD multidisciplinary meeting.7
Until recently, treatment of IPF was limited to lung transplantation in suitable candidates, while the majority of patients could only be offered best supportive or palliative care. However, in 2014, two new antifibrotic medications (nintedanib and pirfenidone) were shown to have a clinically meaningful impact on the natural history of mild to moderate IPF.8,9 Their availability for patients has revolutionised the approach to this once largely untreatable condition.
This article summarises two recently published position statements from the Thoracic Society of Australia and New Zealand and Lung Foundation Australia, outlining the diagnosis10 and treatment of IPF.11 They do not represent guidelines per se but are based on current evidence as well as expert opinion.
For each position statement, a panel of experts contributed to the comprehensive literature review, writing and editing. The statements were independently peer reviewed and endorsed by the Thoracic Society of Australia and New Zealand. All authors had the opportunity to review this summary statement.
Diagnosis
The clinical presentation of IPF is usually insidious and non-specific, resulting in a significant delay from symptom onset to diagnosis.12 Cardinal symptoms include dyspnoea on exertion, non-productive cough, decreased exercise tolerance13 and weight loss.3 Physical examination may reveal bibasal crackles and, in 30–50% of patients, digital clubbing may be present.14,15 It is essential to assess patients thoroughly for other potential causes of fibrotic lung disease, via a comprehensive history and examination (Box 1). Common differential diagnoses include connective tissue disease, drug-induced or occupational lung disease, and hypersensitivity pneumonitis.
High resolution computed tomography has revolutionised the diagnosis of IPF as it allows for detailed assessment of the lung parenchyma, which may eliminate the need for an open lung biopsy.16,17 Based on international guidelines (Box 2), the presence of features consistent with a usual interstitial pneumonia pattern, including subpleural, reticular changes and honeycombing (Box 3), is diagnostic of IPF when other causes for ILD have been excluded.7 In the absence of honeycombing, the predictive value of these features for the presence of usual interstitial pneumonia is reduced and, depending on clinical factors, a surgical lung biopsy may be required to confirm a diagnosis of IPF (Box 4).
Role of the multidisciplinary meeting
Current international guidelines recommend a dynamic multidisciplinary discussion between expert respiratory physicians, radiologists and pathologists as the gold standard for IPF diagnosis.7 Several studies suggest that the use of ILD-specific multidisciplinary meetings leads to greater accuracy and confidence in IPF diagnosis than clinicians working in isolation,18-20 and frequently leads to a change in diagnosis from that proffered by the referring physician.21
Expert ILD multidisciplinary meetings may display considerable heterogeneity in final diagnoses.18,22 To achieve standardisation, based on relatively limited and evolving data supplemented by expert opinion, we have produced a statement10 outlining the conduct of the multidisciplinary meeting regarding core clinical input, discussion and diagnostic outputs.
Data that should be presented at an ILD multidisciplinary meeting are described in Box 5. A detailed and systematic consideration of all diagnostic material is essential to maximise diagnostic confidence from an ILD multidisciplinary meeting. Where data are inadequate for diagnosis to occur, as is common when the history is incomplete or imaging is suboptimal, repeat or further investigation before re-presentation may be required (online Appendix 1).
We suggest that the diagnosis of IPF should take place in a dedicated ILD multidisciplinary meeting. The suggested governance and structure of the multidisciplinary meeting is outlined in online Appendix 2. We recommend that an ILD-dedicated meeting involve at least two respiratory physicians (including the treating physician), a radiologist and a histopathologist, each with ILD expertise, and that the meeting adopt a consensus approach to diagnosis.
Diagnostic outputs from the ILD multidisciplinary meeting should include the final ILD diagnosis, the differential diagnosis and the level of diagnostic certainty. Secondary outputs, such as prediction of disease behaviour as well as treatment goal and management recommendations, may be useful but their value greatly depends on the constitution of the multidisciplinary meeting. Standardised correspondence of the multidisciplinary meeting discussion should be communicated to the treating clinician(s).
As multidisciplinary meetings are complex in their structure and governance, adequate infrastructure and human resource support are vital. In Australia and New Zealand, the location of ILD multidisciplinary meetings may be biased towards larger tertiary centres where core members from various disciplines are more likely to be co-located. Tele- and video-conferencing is an invaluable tool to overcome the geographic separation of ILD experts, enabling access for outer metropolitan, regional and remote centres, as well as for patients who are cared for in the private sector.
Treatment
Until recently, there were no effective therapies for IPF. With the publication of two landmark clinical trials in 2014, antifibrotic therapies have gained widespread approval.8,9 However, the generalisability of these clinical trials is uncertain, especially in the broader IPF disease spectrum.
Despite these advances in pharmacological therapy, IPF remains a progressive and fatal disease. Non-pharmacological interventions including pulmonary rehabilitation, supplemental oxygen, and management of comorbidities and disease-related symptoms are vital to the holistic care of patients with IPF. A suggested treatment algorithm is shown in Box 6.
In phase 3 double-blind randomised clinical trials, pirfenidone8,23 (a pleotrophic novel antifibrotic) and nintedanib9 (an intracellular tyrosine kinase inhibitor) were separately shown to slow disease progression in IPF compared with placebo. In the ASCEND (assessment of pirfenidone to confirm efficacy and safety in IPF) study, there was a 47.9% relative reduction in the proportion of patients who died or had a decline in forced vital capacity (FVC) ≥ 10% at 52 weeks on treatment compared with patients receiving placebo.8 Similarly, in the INPULSIS studies of nintedanib, patients receiving nintedanib had an approximately 50% reduction in annual fall in FVC, with a fall in FVC of 114.7 mL/year and 113.6 mL/year in INPULSIS studies 1 and 2 compared with 239.9 mL/year and 207.3 mL/year in the respective placebo groups.9
In each of these clinical trials, patients had mild to moderate disease, with patients with an FVC < 50% excluded from all trials. The ASCEND study8 of pirfenidone also excluded patients with an FVC > 90%, while the INPULSIS9 studies of nintedanib did not specify an upper FVC limit. Importantly, in post-hoc analyses, patients with very mild disease (FVC > 90% for nintedanib, FVC > 80% for pirfenidone)24,25 had an attenuation of FVC decline similar to that of patients with moderate physiological impairment. However, in severe disease (FVC < 50%), there is very little controlled data to guide treatment. Cohort studies of pirfenidone in real-world populations26,27 suggest that treatment of patients with severe IPF with pirfenidone may be beneficial. A controlled study of nintedanib in patients with severe disease is currently recruiting (NCT02802345).
Other fibrosing lung diseases such as connective tissue disease-related ILD, chronic hypersensitivity pneumonitis and asbestosis may present with a usual interstitial pneumonia pattern on radiology and/or histopathology. A minority of patients (about 10%) remain unclassifiable despite thorough investigation and discussion at an ILD multidisciplinary meeting.28,29 As there is currently no evidence to support the use of antifibrotic medications in patients with non-IPF usual interstitial pneumonia or unclassifiable disease, a clinical trial, if available, should be considered.
The suggestions from the Thoracic Society of Australia and New Zealand and Lung Foundation Australia position statement on IPF treatment for the use of antifibrotic medications are listed in Box 7. The criteria for government-funded IPF treatment in Australia and New Zealand are also summarised in Box 8. In Australia, funding is approved for patients with mild to moderate IPF (FVC ≥ 50% and diffusion capacity of the lungs for carbon monoxide [DLco] ≥ 30%); however, in New Zealand, funding has been restricted to patients with moderate IPF only (FVC ≥ 50% but ≤ 80%, and DLco ≥ 30%).
The main adverse events associated with pirfenidone are gastrointestinal, including nausea, diarrhoea, dyspepsia and vomiting. The other common adverse event is a photosensitive rash, which occurs in 25% of patients. Diarrhoea is the most common adverse event with nintedanib, occurring in about 60% of patients. Other common adverse events are nausea, vomiting and decreased appetite. In most cases, adverse events with both medications are mild and rarely lead to treatment discontinuation. Both antifibrotic medications may cause liver function abnormalities and liver function tests should therefore be regularly monitored.
A summary of suggestions for other pharmacological therapies is provided in Box 7.
Management of comorbidities
Comorbidities are common in patients with IPF and include cardiovascular disease, gastroesophageal reflux disease, pulmonary hypertension and sleep-disordered breathing.2 The optimisation of comorbidities has the potential to prolong and improve quality of life, and therapies to target these diseases should therefore be considered (Box 9).
Gastroesophageal reflux disease
Gastroesophageal reflux disease is highly prevalent in patients with IPF and is thought to contribute to its pathogenesis (through microaspiration resulting in alveolar epithelial injury) or perhaps be exacerbated by IPF (via changes in intrathoracic pressures). While there have been no randomised controlled trials, initial retrospective studies suggested a possible therapeutic benefit of antacid therapy.30,31 However, a more recent post-hoc analysis of the pooled data from the placebo arms of the pirfenidone and nintedanib studies found no benefit and possible increased harm resulting from an increased risk of generalised and respiratory infections with antacid therapy.32
Pulmonary hypertension
The presence of pulmonary hypertension in IPF is associated with increased mortality.33 Despite multiple randomised controlled trials, treatment with pulmonary vasodilators including sildenafil, bosentan, ambrisentan, macitentan and riociguat have failed to demonstrate benefit in people with IPF.34-37 A recent network meta-analysis, however, has suggested that sildenafil may extend survival in selected patients with IPF.38 Therapy with sildenafil could be considered in specific cases in consultation with a pulmonary hypertension specialist centre.
Sleep-disordered breathing
Nocturnal hypoxaemia, with or without apnoea, is common in IPF, with higher nocturnal desaturation events and lower nadir oxygen saturation linked to increased mortality in patients with ILD.39 In a non-randomised study, compliance with continuous positive airway pressure in patients with IPF with moderate to severe sleep apnoea was associated with improved daytime functioning and sleep quality, as well as improved survival at 2 years.40
Combined pulmonary fibrosis and emphysema
Combined pulmonary fibrosis and emphysema refers to the coexistence of pulmonary fibrosis and radiological emphysema. The therapeutic options for patients with combined pulmonary fibrosis and emphysema are limited, as there is a paucity of data on this subset of patients. A post-hoc analysis of patients with emphysema less than the extent of fibrosis and a forced expiratory volume in 1 second/FVC ratio above 80% suggests that treatment with nintedanib confers therapeutic benefit with a similar effect size as for patients without emphysema.41 While there are no data on antifibrotic therapy in patients with emphysema extent greater than fibrosis, standard bronchodilator therapy and supplemental oxygen for those with airflow limitation could be of benefit in selected patients.
Non-pharmacological therapy
Non-pharmacological therapies are important in the management of IPF; patients often have a high burden of symptoms, marked exercise intolerance and poor quality of life, which often persist despite antifibrotic therapy. Early introduction of pulmonary rehabilitation, palliative measures and referral for lung transplantation in selected patients are important, as IPF remains an incurable and fatal disease (Box 10).
Oxygen
Resting oxygen requirements, as well as the degree of sleep and exercise-induced hypoxaemia, have been associated with adverse outcomes in IPF; however, it is unknown whether prevention of hypoxaemia assuages this risk. Currently, recommendations for supplemental oxygen in IPF have been extrapolated from trials in chronic obstructive pulmonary disease.42,43 In Australia and New Zealand, oxygen therapy is suggested for patients with resting hypoxaemia (Pao2 < 55 mmHg, or < 60 mmHg in the presence of end-organ damage). Nocturnal oxygen can be considered in patients who desaturate below 88% for at least one-third of total sleep time, and intermittent ambulatory oxygen may be considered in hypoxaemic individuals who have improvement in exercise capacity or dyspnoea with oxygen therapy. However, evidence to support this approach is equivocal, and factors such as cost, physical challenges of transporting devices and the limited duration of oxygen cylinders should be considered when suggesting oxygen therapy, particularly in the ambulatory setting.
Pulmonary rehabilitation
There is growing evidence that pulmonary rehabilitation has important effects on symptoms, functional capacity and wellbeing in patients with IPF. A recent Cochrane review showed that an 8–12-week pulmonary rehabilitation program, including whole body endurance and resistance training as well as education and psychosocial support, led to improvements in 6-minute walk distance, dyspnoea and health-related quality of life. However, gains in exercise capacity often diminish over 6–12 months.44 Patients with milder disease appear to have more sustained benefit and early referral should therefore be considered.45
Lung transplantation
Lung transplantation remains the only definitive cure for IPF, and it is recommended that appropriate patients be referred at the time of IPF diagnosis, regardless of disease severity.46 This recommendation reflects the phenotypic heterogeneity and thus difficulties of predicting the individual course. The overall prognosis for IPF is poor compared with other indications for lung transplantation, illustrated by the fact that IPF has the greatest waiting list mortality, despite the shortest median waiting list time.47
Chronological age is no longer an absolute contraindication to transplant; rather, the comorbidities with end-organ damage and physiological reserve are the main criteria used. Indications for lung transplant listing include desaturation to < 88% or distance < 250 m on 6-minute walk testing or > 50 m decline in 6-minute walk distance over a 6-month period, a fall in FVC ≥ 10% and/or in DLco ≥ 15% during 6-month follow-up, the presence of pulmonary hypertension, or hospitalisation for acute exacerbation, respiratory decline or pneumothorax.46
Palliative care
Palliative care is an integrated, dynamic and individualised process which should be addressed at all stages of disease, with goals shifting to meet the changing needs of patients. Palliative care focuses on symptom management, advance care directives and end-of-life planning, aiming to improve the quality of life of patients and their families.
The use of oral opioids to reduce dyspnoea, a common and debilitating symptom in IPF, has been reported; however, there is limited evidence for the use of benzodiazepine or oxygen. While opioids and over-the-counter cough suppressants have been used to treat cough in IPF, there is little evidence that these measures are effective, and a thorough search for reversible causes of cough should be undertaken in all patients with IPF.
Conclusion
It is a promising time in the history of IPF, with advances in both the diagnostic process and management of this condition. Accurately diagnosing IPF is critically important, given the positive outcomes with correct use of antifibrotic therapies in a subgroup of patients. The next decade will see further evolution such as advances in high resolution computed tomography technology, newer methods of sampling lung tissue, biomarkers and genetic testing, which will refine the multidisciplinary meeting diagnostic process, improve our ability to provide prognosis for patients, and further optimise and personalise management pathways.
Box 1 – Clinical features and investigation findings in idiopathic pulmonary fibrosis
History- Older age (median age, 66 years)
- Exertional dyspnoea (present in 90% of patients)13
- Non-productive cough (present in > 70% of patients)13
- Absence of features suggestive of other cause of interstitial lung disease including clinical features of connective tissue disease, occupational and recreational causes and medication related
- Finger clubbing (present in 40–65% of patients)15
- Bibasilar crackles
- Features of cor pulmonale (in advanced disease)
- Restrictive ventilatory deficit on spirometry
- Reduction in diffusion capacity
- Six-minute walk test with oximetry may demonstrate reduction in walk distance and oxygen desaturation
- High resolution computed tomography: usual interstitial pneumonia pattern of fibrosis
- Surgical lung biopsy (if required): usual interstitial pneumonia pattern of fibrosis
- Serum and bronchoalveolar lavage biomarkers and genetic testing is not routinely available for clinical practice in Australia
Box 2 – High resolution computed tomography criteria for diagnosing usual interstitial pneumonia (UIP) pattern*
|
UIP pattern (all four features) |
Possible UIP pattern (all three features) |
Inconsistent with UIP pattern (any of seven features) |
|||||||||||||
|
|
|||||||||||||||
|
Subpleural, basal predominance |
Subpleural, basal predominance |
Upper and mid lung predominance |
|||||||||||||
|
|
|||||||||||||||
|
* Adapted from Raghu.7 |
|||||||||||||||
Box 3 – Radiological features of usual interstitial pneumonia
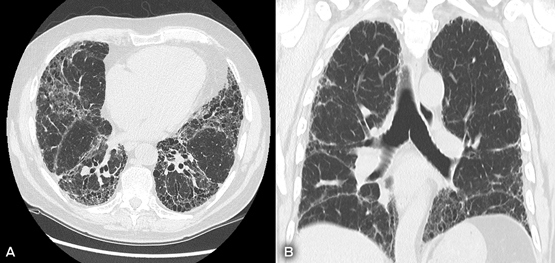
High resolution computed tomography scan showing reticular abnormality and honeycombing, and (A) subpleural predominance (axial view) and (B) apicobasal predominance (coronal view).
Box 4 – Histopathological features of usual interstitial pneumonia
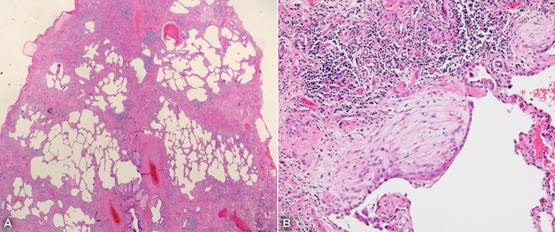
Surgical lung biopsy showing (A) gross architectural destruction of lung parenchyma and (B) honeycombing with fibroblastic foci (haemotoxylin–eosin stain).
Box 5 – Core and ancillary data to be presented in a standardised format at an interstitial lung disease multidisciplinary meeting∗
|
Core data (to be presented in every case) |
Ancillary data (to be presented when available) |
||||||||||||||
|
|
|||||||||||||||
|
Clinical history
|
Investigations
|
||||||||||||||
|
|
|||||||||||||||
|
∗ Adapted from Prasad et al.10 |
|||||||||||||||
Box 6 – Suggested treatment algorithm for idiopathic pulmonary fibrosis (IPF)∗
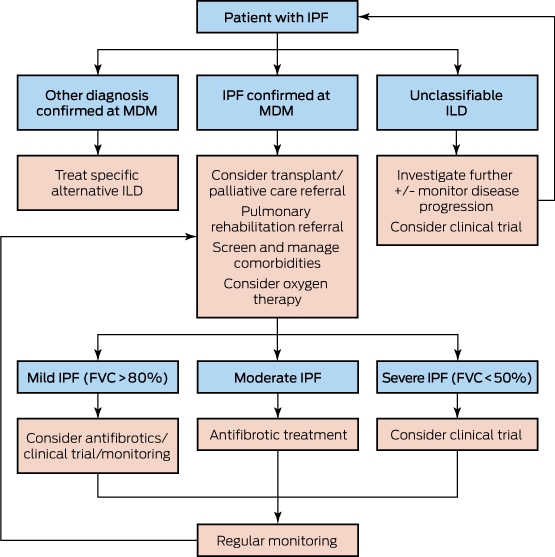
FVC = forced vital capacity; ILD = interstitial lung disease; MDM = multidisciplinary meeting. ∗ Adapted from Jo et al.11
Box 7 – Pharmacological therapies for idiopathic pulmonary fibrosis (IPF)11
|
|
|||||||||||||||
|
Antifibrotic therapy
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
Box 8 – Criteria for funded treatment with antifibrotic therapy in Australia and New Zealand∗
|
Australia (PBS) |
New Zealand (PHARMAC)† |
||||||||||||||
|
|
|||||||||||||||
|
Multidisciplinary diagnosis of IPF |
Physician diagnosis of IPF, confirmed by CT or biopsy |
||||||||||||||
|
High resolution CT consistent with usual interstitial pneumonia within 12 months |
FVC, 50–80% |
||||||||||||||
|
FVC ≥ 50% |
Funding discontinued if disease progresses (fall in FVC ≥ 10% in 12 months) |
||||||||||||||
|
FEV1/FVC ratio > 0.7 |
|
||||||||||||||
|
DLCO ≥ 30% |
|
||||||||||||||
|
Interstitial lung disease not due to other known cause |
|
||||||||||||||
|
|
|||||||||||||||
|
CT = computed tomography; DLco = diffusion capacity of the lungs for carbon monoxide; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; IPF = idiopathic pulmonary fibrosis. PBS = Pharmaceutical Benefits Scheme; PHARMAC = Pharmaceutical Management Agency; * Adapted from Jo et al.11 † Has only approved pirfenidone. |
|||||||||||||||
Box 9 – Management of comorbidities for idiopathic pulmonary fibrosis11
Gastroesophageal reflux disease- It is unclear whether the treatment of gastroesophageal reflux disease with antacid therapy confers benefit in idiopathic pulmonary fibrosis and thus treatment should be considered on an individual basis until further data are available
- Treatment with pulmonary vasodilators is currently not supported by controlled data
- Polysomnography could be considered if obstructive sleep apnoea or nocturnal hypoxia is suspected
- Supplemental oxygen and inhaled bronchodilator therapy may alleviate breathlessness in some patients with combined pulmonary fibrosis and emphysema
- In a subset of patients with fibrosis greater than emphysema without airway obstruction, antifibrotic therapy can be considered
Box 10 – Non-pharmacological management of idiopathic pulmonary fibrosis11
Oxygen therapy- Long term oxygen therapy is suggested in patients with resting hypoxaemia
- Supplemental oxygen may be considered in patients with significant exercise and sleep-related oxygen desaturation
- Early referral to pulmonary rehabilitation is suggested, especially in those with milder disease as it confers meaningful benefits in exercise capacity, symptoms and health-related quality of life
- Early referral to a lung transplant centre is suggested for potentially eligible patients
- Early palliative/supportive care may be helpful in managing debilitating symptoms of dyspnoea, cough and fatigue
- Advance care planning should be offered early, when patients are relatively well to ensure that their wishes and those of their families can be heard and respected
Competing interests
For competing interest declarations, see online .
Acknowledgements
David Beatson was a well known New Zealand journalist, broadcaster and editor. Among his many roles, he served as Prime Minister’s chief press officer, and chair of NZ on Air. A thoughtful, erudite and engaging patient, David died from IPF on 21 September 2017. We acknowledge his important contribution as patient author on this article.
References
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 431-440.
- Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J 2015; 46: 1113-1130.
- Kim HJ, Perlman D, Tomic R. Natural history of idiopathic pulmonary fibrosis. Respir Med 2015; 109: 661-670.
- Aggarwal R, McBurney C, Schneider F, et al. Myositis-associated usual interstitial pneumonia has a better survival than idiopathic pulmonary fibrosis. Rheumatology (Oxford) 2017; 56: 384-389.
- Miyazaki Y, Tateishi T, Akashi T, et al. Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest 2008; 134: 1265-1270.
- Lake F, Proudman S. Rheumatoid arthritis and lung disease: from mechanisms to a practical approach. Semin Respir Crit Care Med 2014; 35: 222-238.
- Raghu G. An Official ATS/ERS/JRS/ALAT Statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Crit Care Med 2011; 183: 788-824.
- King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083-2092.
- Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071-2082.
- Prasad JD, Mahar A, Bleasel J, et al. The interstitial lung disease multidisciplinary meeting: a position statement from the Thoracic Society of Australia and New Zealand and the Lung Foundation Australia. Respirology 2017; 22: 1459-1472.
- Jo HE, Troy LK, Keir G, et al. Treatment of idiopathic pulmonary fibrosis in Australia and New Zealand: a position statement from the Thoracic Society of Australia and New Zealand and the Lung Foundation Australia. Respirology 2017; 22: 1436-1458.
- Sgalla G, Biffi A, Richeldi L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology 2016; 21: 427-437.
- Lee AS. The burden of idiopathic pulmonary fibrosis: an unmet public health need. Respir Med 2014; 108: 955-967.
- Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006; 3: 285-292.
- Sarkar M, Mahesh DM, Madabhavi I. Digital clubbing. Lung India 2012; 29: 354-362.
- Grenier P, Valeyre D, Cluzel P, et al. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179: 123-132.
- Swensen SJ, Aughenbaugh GL, Myers JL. Diffuse lung disease: diagnostic accuracy of CT in patients undergoing surgical biopsy of the lung. Radiology 1997; 205: 229-234.
- Flaherty KR, King TE, Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170: 904-910.
- Flaherty KR, Andrei AC, King TE Jr, et al. Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis? Am J Respir Crit Care Med 2007; 175: 1054-1060.
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4: 557-565.
- Jo HE, Glaspole IN, Levin KC, et al. Clinical impact of the interstitial lung disease multidisciplinary service. Respirology 2016; 21: 1438-1444.
- Walsh SLF, Hansell D, Flaherty KR, et al. Accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: a study of 275 pulmonologists from 54 countries [abstract]. Am J Respir Crit Care Med 2017; 195: A1130.
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377: 1760-1769.
- Albera C, Bradford WZ, Costabel U, et al. Pirfenidone is efficacious in patients with idiopathic pulmonary fibrosis (IPF) and mild restrictive disease [abstract]. Am J Respir Crit Care Med 2015; 191: A1018.
- Kolb M, Richeldi L, Behr J, Maher TM. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. 2017; 72: 340-346.
- Sakamoto S, Itoh T, Muramatsu Y, et al. Efficacy of pirfenidone in patients with advanced-stage idiopathic pulmonary fibrosis. Intern Med 2013; 52: 2495-2501.
- Okuda R, Hagiwara E, Baba T, et al. Safety and efficacy of pirfenidone in idiopathic pulmonary fibrosis in clinical practice. Respir Med 2013; 107: 1431-1437.
- Troy L, Glaspole I, Goh N, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J 2014; 43: 1529-1530.
- Ryerson CJ, Urbania TH, Richeldi L, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J 2013; 42: 750-757.
- Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 184: 1390-1394.
- Lee JS, Collard HR, Anstrom KJ, et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med 2013; 1: 369-376.
- Raghu G, Crestani B, Bailes Z, et al. Effect of anti-acid medication on reduction in FVC decline with nintedanib. Eur Respir J 2015; 46 (Suppl 59): OA4502.
- Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest 2007; 132: 998-1006.
- Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med 2013; 158: 641-649.
- Raghu G, Million-Rousseau R, Morganti A, et al. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J 2013; 42: 1622-1632.
- King TE Jr. Bosentan for idiopathic pulmonary fibrosis. Curr Opin Investig Drugs 2008; 9: 1171-1179.
- Zisman DA, Schwarz M, Anstrom KJ, et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med 2010; 363: 620-628.
- Rochwerg B, Neupane B, Zhang Y, et al. Treatment of idiopathic pulmonary fibrosis: a network meta-analysis. BMC Med 2016; 14: 18.
- Kolilekas L, Manali E, Vlami KA, et al. Sleep oxygen desaturation predicts survival in idiopathic pulmonary fibrosis. J Clin Sleep Med 2013; 9: 593-601.
- Pihtili A, Bingol Z, Kiyan E, et al. Obstructive sleep apnea is common in patients with interstitial lung disease. Sleep Breathing 2013; 17: 1281-1288.
- Corte T, Cottin V, Taniguchi H, et al. Effect of baseline emphysema on reduction in FVC decline with nintedanib in the INPULSIS trials. Respirology 2015; 20: 57.
- Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working Party. Lancet 1981; 317: 681-686.
- Nocturnal Oxygen Therapy Trial Group. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Ann Intern Med 1980; 93: 391-398.
- Dowman L, Hill CJ, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst Rev 2014; 10: CD006322.
- Holland AE, Hill CJ, Glaspole I, et al. Predictors of benefit following pulmonary rehabilitation for interstitial lung disease. Respir Med 2012; 106: 429-435.
- Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014 – an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2015; 34: 1-15.
- Bennett D, Fossi A, Bargagli E, et al. Mortality on the waiting list for lung transplantation in patients with idiopathic pulmonary fibrosis: a single-centre experience. Lung 2015; 193: 677-681.
Linked content
-
MJA InSight: Idiopathic pulmonary fibrosis: promise on the horizon
Provenance: Not commissioned; externally peer reviewed.