





Volume 190 - Issue 4 Supplement
Are common childhood or adolescent infections risk factors for schizophrenia and other psychotic disorders?
Authors: Ian B Hickie, Richard Banati, Claire H Stewart and Andrew R Lloyd
Med J Aust 2009; 190 (4 Suppl): S17. || doi: 10.5694/j.1326-5377.2009.tb02369.x
Published online: 16 February 2009
Published online: 16 February 2009
Abstract
Postnatal infection may represent a preventable risk factor for onset of psychotic disorders in adolescence and early adulthood.
The mechanism of action is likely to involve site-directed triggering of the brain’s innate immune system, mediated principally through localised activation of microglial cells. This triggering may occur in response to systemic inflammatory stimuli, without direct involvement of the central nervous system.
Microglial activation can represent a primary response or a secondary phenomenon at sites made vulnerable by prior injury; that is, areas containing previously activated microglia will respond more strongly to a new stimulus.
The presence of activated microglia is indicative of a recent insult or active disease. It is not characteristic of long-established neurodevelopmental abnormalities.
Activated microglia, acting through a variety of cytokine and other signal systems, have the capacity to significantly interfere with synaptic turnover and thus, over time, alter synaptic architecture and function.
This pathophysiological path should be investigated more systematically as it may explain a novel “neuroprotective” mode of action for some existing antipsychotic compounds.
Simple genetic theories of schizophrenia and other psychotic disorders are now being challenged by more complex models of gene–environment interactions.1 These gene–environment models strive not only to identify significant environmental risks, but also to assess their relationship with the onset of these disorders in adolescence (see Hickie and McGorry,2 page S5; McGorry and colleagues,3 page S33 in this supplement).1,4 Critically, gene–environment models shift the focus of preventive and early-intervention research away from a narrow neurodevelopmental focus, towards potential reduction of major environmental risks.1
Traditionally, the postpubertal onset of schizophrenia, psychotic disorders and major mood disorders has been thought to result from the effect of normal developmental factors (eg, hormonal factors, frontal lobe maturation, synaptic pruning) on those who already carry some pre-existing genetic, environmental (eg, intrauterine infection) or neurodevelopmental vulnerability.5 More recently, this view has been challenged by the concept of a “double hit”, whereby exposure to one or more additional risk factors in adolescence (eg, misuse of cannabis or amphetamines and other stimulants [see Hermens and colleagues,6 page S22; Wood and colleagues,7 page S10], head injury, brain infection, emotional trauma) precipitates the progression from being “at risk” to an active illness state.
In this article, we focus on the possible role of postnatal or postpubertal infections (or other inflammatory stimuli) as discrete risk factors. We do not assume that such infective agents are rare, neurotropic or neurotoxic. Indeed, such infections are not necessarily directly involved with the central nervous system (CNS) or associated with clinical evidence of CNS involvement. The way in which systemic infections or inflammation can give rise to a wide range of CNS-related symptoms,8 and that these phenomena persist beyond the acute illness phase, is increasingly recognised.9
Our focus on exposure to infective stimuli is driven by:
epidemiological studies linking major psychiatric disorders with infective exposures (albeit often dependent on host genotype; see Box 1);
altered immune function in people with schizophrenia that may indicate an ongoing inflammatory process;22-24
evidence that common infective stimuli outside the CNS modify the responses of the brain’s innate immune system (eg, microglial activation occurs in the presence of peripheral immune activation25-27);
clinical evidence that recent infective stimuli are correlated with a range of short- or longer-term neurological, cognitive and other neuropsychiatric syndromes;9 and
neuroimaging data indicative of microglial activation (MA) in persons with schizophrenia (see Banati and Hickie,28 page S26).29
Microglial response to peripheral inflammation or infection
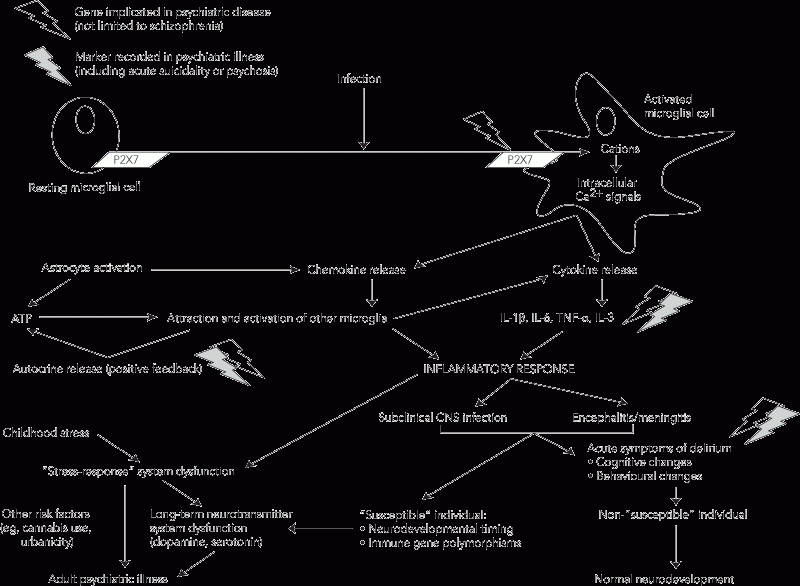
Microglia, unlike other glial cells, are not of neuroectodermal origin. They are derived from circulating monocytes, and are continuously recruited into the brain (at a low rate), where they undergo differentiation in situ. When activated by one of a range of insults,30-32 microglia release pro-inflammatory cytokines and nitric oxide (NO). MA has been reported across a broad range of neuroinflammatory and neurodegenerative disorders.33-36 Importantly, microglial dysfunction has been proposed as a mechanism linking in-utero viral exposure with increased risk of schizophrenia.37
We offer a different interpretation of the importance of detection of MA in adults with schizophrenia or other psychotic disorders. Key additional observations include that MA is transient, not present in old gliotic scars, and does not occur in response to stable neurodevelopmental abnormalities.33-36 That is, MA implies a recent, active or ongoing pathophysiology and is, therefore, unlikely to represent a response to an in-utero exposure alone. After cessation of a pathological stimulus, activated microglia persist and continue to affect synaptic function. In animal models, the minimal period for MA is 1 month. In the adult human brain, the disappearance of activated microglia may be significantly slower because of a protracted pathological stimulus or the effects of other adaptive processes, such as glial cell regeneration.38,39
In addition to their immunomodulatory role, it has recently emerged that microglia are vital members of the “quadpartite synapse”, which also includes presynaptic and postsynaptic neurones, and astrocytes.40 Their release of pro-inflammatory cytokines (interleukin [IL]-1β, IL-6 and tumour necrosis factor [TNF]-α) influences neurotransmission, potentially in the long term, through positive feedback loops. This bridging role of MA that links immune response and altered neurotransmission suggests a plausible pathway from infection to persistent neuropsychiatric disorders. Of particular interest is the observation that MA may be triggered by a single exposure to pathogen-associated molecular patterns, such as the lipopolysaccharide component of the cell wall of gram-negative bacteria.25 These stimuli interact with pattern recognition receptors, such as toll-like receptors, to induce TNF-α production and associated neuronal injury. These adverse effects may then be sustained for many months in the absence of systemic inflammation.25
Communication between microglia is coordinated by purinergic signalling, including the ionotropic ATP receptor, P2X7. Polymorphisms of this receptor (eg, gln460arg) have recently been associated with depression and bipolar disorder.41,42 Such receptor polymorphisms may confer dysfunctional signalling, influencing long-term potentiation,43 and thereby alter synaptic communication. We do not suggest that immune activation alone induces psychotic symptomatology. Instead, we propose that exposure to infection is a potential priming event for the longer-term development of abnormal signalling patterns that underpin such symptoms.
The tentative findings of MA in schizophrenia28,29 are consistent with previous structural imaging studies demonstrating active brain changes during the period of onset of psychotic symptoms.44,45 Calprotectin, a novel inflammatory marker protein, was found to be localised to microglia and elevated in postmortem frontal cortex tissue from people with schizophrenia and other psychotic disorders.46 Further, a relationship between the degree of MA and the extent of neurophysiological impairment (as measured by changes in cortical evoked potentials) implies that MA is linked to the course of structural brain change and illness progression in people with schizophrenia.28 It has also been suggested recently that suicidal behaviour in people with schizophrenia or depression may be linked with MA.47
Site-directed vulnerability within the CNS
Central to our postnatal infective hypothesis is evidence that brain tissue containing activated microglia will be a preferred site for recruitment of other bloodborne immunological cells, such as activated T lymphocytes. This site-directed homing of peripheral leucocytes into brain regions containing activated microglia has been demonstrated in models of optic nerve transaction48 and in the facial nucleus after injury to the peripheral facial nerve.49,50 In these models, T lymphocytes infiltrate the CNS through an intact blood–brain barrier and interact with microglia to modulate the brain’s neural response.51,52
Consequently, we propose a model for the development of major psychiatric disorders in which activated microglia represent a key link between postnatal infection and later psychopathological events (Box 2). In this model, changes in the state of the brain’s innate immune system (notably microglia):
Timing of infective exposures
To date, the most plausible evidence for infection as a major risk factor for schizophrenia has been obtained from epidemiological studies53 and animal models of intrauterine exposure.54 These models are consistent with epidemiological evidence linking prenatal infection with increased risks of most major neurological and psychiatric disorders, including (but not limited to) schizophrenia and other psychotic disorders.55 In our view, however, the plausibility of infective stimuli as important risk factors has also been constrained by a preoccupation with fetal development. Although neurogenesis occurs largely before birth, other key developmental mechanisms such as synaptogenesis, synaptic pruning and myelination continue throughout childhood and adolescence. Hence, the potential adverse impacts of infective or inflammatory stimuli continue up to and include the typical age of onset of these disorders — between 15 and 25 years.
A range of childhood infections have been linked to adult psychotic disorders.10,56 As early childhood is associated with the highest rates of gastroenteritis and common respiratory infections, these exposures may be relevant in those predisposed to the CNS consequences of peripheral infection or inflammation. Adolescence is again an important time for infectious exposures, as it is a high-risk period for exposure to both neurotropic and systemic viral pathogens, including herpes simplex virus type 1 (HSV1) and HSV2, Epstein–Barr virus and cytomegalovirus.
The role of genes and viral exposures in risk of illness and clinical phenotypes
Preliminary evidence of interactions between at-risk genes, infective risk factors, phenotypes and biological correlates of psychotic disorders has recently appeared. Although presence of the val158met catechol-O-methyltransferase polymorphism and HSV1 exposure both increase the risk of cognitive changes, the combination of these two factors is associated with an 85-fold increased risk of cognitive impairment in patients with bipolar disorder.17 Prasad and colleagues reported reduced prefrontal grey matter among HSV1-exposed first-episode patients with schizophrenia.57 Prasad and colleagues also examined the impact of a polymorphism of major histocompatibility complex class I polypeptide-related sequence B (MICB) on grey matter changes.58 Critically, and suggestive of a genuine gene–environment interaction, they reported that MICB genotype and HSV1 exposure both contributed to greater grey matter differences between patient and control subjects. Shirts and colleagues have also suggested that, not only are there specific associations between genes that regulate IL-18 pathways and schizophrenia, but that some specific single-nucleotide polymorphisms are also associated with elevated HSV1 antibody titres.24 It is also becoming increasingly evident that the severity of acute sickness phenomena (presumably reflecting both degree of immune activation and CNS-related phenomena) after common infective stimuli is itself dependent on host genotype.59
Ongoing immune dysfunction in patients with schizophrenia
These speculative views are supported by findings that adult patients with schizophrenia demonstrate evidence of altered immune function (eg, in-vivo increases in IL-1RA [receptor agonist], soluble IL-2R and IL-6 production and an in-vitro decrease in IL-2 production) that is consistent with ongoing inflammatory responses.60,61 Similar findings of ongoing immune disturbance have been noted in other major psychiatric disorders, particularly affective disorders.62-64 Evidence that genetic variations in immune response are also susceptibility factors for major psychiatric disorders (eg, P2X7 and risk to bipolar disorder and unipolar depression) suggests that progress in our understanding of these complex relationships may also depend on more sophisticated human and animal model designs that capture relevant gene–environment interactions.
Immunomodulatory effects of treatments
Recently, there has been some interest in two novel therapeutic strategies. First, some antipsychotic agents may have immunomodulatory properties — perospirone, quetiapine, aripiprazole, risperidone and, to some extent, ziprasidone have been shown to inhibit release of pro-inflammatory cytokines or NO from activated microglia.65,66 Further, olanzapine (but not clozapine or haloperidol) has been shown to inhibit NO release from mouse microglia.67 Second, anti-infective agents that have anti-inflammatory properties (eg, minocycline) may be used to treat psychotic disorders.68
Although these approaches may be consistent with the hypotheses we have outlined in this article, they are clearly highly speculative. In fact, a common element of many recently proposed adjunctive “neuroprotective” strategies (eg, lithium carbonate, omega-3 fatty acids) may be their capacity to moderate MA that occurs in response to a wide range of pathological stimuli.3
Conclusion
This article has focused on the potential role of common postnatal infections as risk factors for the adolescent onset of schizophrenia and other psychotic disorders. Mechanistically, this is assumed to depend on MA as part of an augmented and site-specific reaction of the brain’s innate immune system. This could be in response to a single severe infection (presumably rarely) or, more commonly, through aggravation of pre-existing and localised microglial reaction. Importantly, persistent MA is associated with disrupted neural circuits and, consequently, dysfunctional neurochemistry. Specifically, we also propose that the adverse effects of common postnatal infections depend on interactions with key immunomodulatory genes (eg, P2X7, MICB), priming infections or other earlier CNS insults. This line of research opens new pathways for both preventive and early intervention strategies that utilise either existing or novel pharmacological strategies.
Competing interests
None identified.
References
- Caspi A, Moffitt TE. Gene–environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci 2006; 7: 583-590. 0_i1092133
- Hickie IB, McGorry PD. Characterising novel pathways to schizophrenia [editorial]. Med J Aust 2009; 190 (4 Suppl): S5-S6. 0_i1092135
- McGorry PD, Yung AR, Pantelis C, Hickie IB. A clinical trials agenda for testing interventions in earlier stages of psychotic disorders. Med J Aust 2009; 190 (4 Suppl): S33-S36. 0_i1092137
- Pantelis C, Yücel M, Wood SJ, et al. Structural brain imaging evidence for multiple pathological processes at different stages of brain development in schizophrenia. Schizophr Bull 2005; 31: 672-696. 0_i1092139
- Paus T, Keshavan M, Giedd JN. Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci 2008; 9: 947-957. 0_i1092141
- Hermens DF, Lubman DI, Ward PB, et al. Amphetamine psychosis: a model for studying the onset and course of psychosis. Med J Aust 2009; 190 (4 Suppl): S22-S25. 0_i1092143
- Wood SJ, Pantelis C, Yung AR, et al. Brain changes during the onset of schizophrenia: implications for neurodevelopmental theories. Med J Aust 2009; 190 (4 Suppl): S10-S13. 0_i1092145
- Hickie I, Hickie C, Bennett B, et al. Biochemical correlates of in vivo cell-mediated immune dysfunction in patients with depression: a preliminary report. Int J Immunopharmacol 1995; 17: 685-690. 0_i1092147
- Hickie I, Davenport T, Wakefield D, et al. Post-infective and chronic fatigue syndromes precipitated by viral and non-viral pathogens: prospective cohort study. BMJ 2006; 333: 575. 0_i1092149
- Rantakallio P, Jones P, Moring J, Von Wendt L. Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int J Epidemiol 1997; 26: 837-843. 0_i1092151
- Leask SJ, Done DJ, Crow TJ. Adult psychosis, common childhood infections and neurological soft signs in a national birth cohort. Br J Psychiatry 2002; 181: 387-392. 0_i1092153
- Koponen H, Rantakallio P, Veijola J, et al. Childhood central nervous system infections and risk for schizophrenia. Eur Arch Psychiatry Clin Neurosci 2004; 254: 9-13. 0_i1092155
- Dalman C, Allebeck P, Gunnell D, et al. Infections in the CNS during childhood and the risk of subsequent psychotic illness: a cohort study of more than one million Swedish subjects. Am J Psychiatry 2008; 165: 59-65. 0_i1092157
- Abrahao AL, Focaccia R, Gattaz W. Childhood meningitis increases the risk for adult schizophrenia. World J Biol Psychiatry 2005; 6 Suppl 2: 44-48. 0_i1092159
- Amminger GP, McGorry PD, Berger GE, et al. Antibodies to infectious agents in individuals at ultra-high risk for psychosis. Biol Psychiatry 2007; 61: 1215-1217. 0_i1092161
- Leweke, FM, Gerth CW, Koethe D, et al. Antibodies to infectious agents in individuals with recent onset schizophrenia. Eur Arch Psychiatry Clin Neurosci 2004; 254: 4-8. 0_i1092163
- Dickerson FB, Boronow JJ, Stallings C, et al. The catechol O-methyltransferase Vall58Met polymorphism and herpes simplex virus type 1 infection are risk factors for cognitive impairment in bipolar disorder: additive gene–environmental effects in a complex human psychiatric disorder. Bipolar Disord 2006; 8: 124-132. 0_i1092165
- Dickerson F, Boronow J, Stallings C, et al. Association of serum antibodies to herpes simplex virus 1 with cognitive deficits in individuals with schizophrenia. Arch Gen Psychiatry 2003; 60: 466-472. 0_i1092167
- Buka SL, Cannon TD, Torrey EF, et al. Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biol Psychiatry 2008; 63: 809-815. 0_i1092169
- Torrey EF, Bartko JJ, Lun ZR, Yolken RH. Antibodies to Toxoplasma gondii in patients with schizophrenia: a meta-analysis. Schizophr Bull 2007; 33: 729-736. 0_i1092171
- Yolken RH, Torrey EF. Infectious agents and gene–environmental interactions in the etiopathogenesis of schizophrenia. Clin Neurosci Res 2006; 6: 97-109. 0_i1092174
- Potvin S, Stip E, Sepehry AA, et al. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry 2008; 63: 801-808. 0_i1092176
- Eaton WW, Byrne M, Ewald H, et al. Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am J Psychiatry 2006; 163: 521-528. 0_pgfId-1100852
- Shirts BH, Wood J, Yolken RH, Nimgaonkar VL. Comprehensive evaluation of positional candidates in the IL-18 pathway reveals suggestive associations with schizophrenia and herpes virus seropositivity. Am J Med Genet B Neuropsychiatr Genet 2008; 147: 343-350. 0_i1092179
- Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007; 55: 453-462. 0_i1092181
- Cagnin A, Taylor-Robinson SD, Forton DM, Banati RB. In vivo imaging of cerebral “peripheral benzodiazepine binding sites” in patients with hepatic encephalopathy. Gut 2006; 55: 547-553. 0_pgfId-1100875
- Banati RB. Neuropathological imaging: in vivo detection of glial activation as a measure of disease and adaptive change in the brain. Br Med Bull 2003; 65: 121-131. 0_i1092184
- Banati R, Hickie IB. Therapeutic signposts: using biomarkers to guide better treatment of schizophrenia and other psychotic disorders. Med J Aust 2009; 190 (4 Suppl): S26-S32. 0_i1092186
- van Berckel BN, Bossong MG, Boellaard R, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry 2008; 64: 820-822. 0_i1092188
- Carpentier P, Duncan D, Miller S. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun 2008; 22: 140-147. 0_i1092190
- Raivich G, Banati RB. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Brain Res Rev 2004; 46: 261-281. 0_pgfId-1100917
- Banati RB, Graeber MB. Surveillance, intervention and cytotoxicity: is there a protective role of microglia? Dev Neurosci 1994; 16: 114-127. 0_i1092193
- Cagnin A, Brooks D, Kennedy A, et al. In-vivo measurement of activated microglia in dementia. Lancet 2001; 358: 461-467. 0_i1092195
- Banati RB. Visualising microglial activation in vivo. Glia 2002; 40: 206-217. 0_i1092197
- Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord 1998; 13: 221-227. 0_pgfId-1100948
- Banati RB, Newcombe J, Gunn RN, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain 2000; 123: 2321-2337. 0_i1092200
- Munn NA. Microglia dysfunction in schizophrenia: an integrative theory. Med Hypotheses 2000; 54: 198-202. 0_CBBIECDA
- Graeber MB, Bise K, Mehraein P. Synaptic stripping in the human facial nucleus. Acta Neuropathol 1993; 86: 179-181. 0_i1092203
- Cagnin A, Myers R, Gunn RN, et al. In vivo visualization of activated glia by [11C] (R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain 2001; 124: 2014-2027. 0_i1092205
- Bennett M. Synaptic P2X7 receptor regenerative-loop hypothesis for depression. Aust N Z J Psychiatry 2007; 41: 563-571. 0_i1092207
- Barden N, Harvey M, Gagne B, et al. Analysis of single nucleotide polymorphisms in genes in the chromosome 12Q24.31 region points to P2RX7 as a susceptibility gene to bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet 2006; 141B: 374-382. 0_i1092209
- Lucae S, Salyakina D, Barden N, et al. P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum Mol Genet 2006; 15: 2438-2445. 0_i1092211
- Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFalpha. Science 2002; 295: 2282-2285. 0_i1092213
- Pantelis C, Velakoulis D, McGorry PD, et al. Neuroanatomical abnormalities before and after onset of psychosis: a cross-sectional and longitudinal MRI study. Lancet 2003; 361: 281-288. 0_CBBBGGAF
- Wood SJ, Pantelis C, Velakoulis D, et al. Progressive changes in the development toward schizophrenia: studies in subjects at increased symptomatic risk. Schizophr Bull 2008; 34: 322-329. 0_CBBIFDAE
- Foster R, Kandanearatchi A, Beasley C, et al. Calprotectin in microglia from frontal cortex is up-regulated in schizophrenia: evidence for an inflammatory process? Eur J Neurosci 2006; 24: 3561-3566. 0_CBBFAHFA
- Steiner J, Bielau H, Brisch R, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 2008; 42: 151-157. 0_i1092218
- Molleston MC, Thomas ML, Hickey WF. Novel major histocompatibility complex expression by microglia and site-specific experimental allergic encephalomyelitis lesions in the rat central nervous system after optic nerve transection. Adv Neurol 1993; 59: 337-348. 0_i1092220
- Moran LB, Graeber MB. The facial nerve axotomy model. Brain Res Brain Res Rev 2004; 44: 154-178. 0_i1092222
- Raivich G, Jones LL, Kloss CU, et al. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci 1998; 18: 5804-5816. 0_i1092224
- Byram SC, Carson MJ, DeBoy CA, et al. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci 2004; 24: 4333-4339. 0_i1092226
- Petitto JM, Huang Z, Lo J, et al. IL-2 gene knockout affects T lymphocyte trafficking and the microglial response to regenerating facial motor neurons. J Neuroimmunol 2003; 134: 95-103. 0_i1092228
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull 2006; 32: 200-202. 0_i1092230
- Rehn AE, Rees SM. Investigating the neurodevelopmental hypothesis of schizophrenia. Clin Exp Pharmacol Physiol 2005; 32: 687-696. 0_i1092232
- Verdoux H. Perinatal risk factors for schizophrenia: how specific are they? Curr Psychiatry Rep 2004; 6: 162-167. 0_i1092234
- Gattaz WF, Abrahao AL, Foccacia R. Childhood meningitis, brain maturation and the risk of psychosis. Eur Arch Psychiatry Clin Neurosci 2004; 254: 23-26. 0_i1092236
- Prasad KM, Shirts BH, Yolken RH, et al. Brain morphological changes associated with exposure to HSV1 in first-episode schizophrenia. Mol Psychiatry 2007; 12: 105-113. 0_i1092238
- Prasad K, Shirts BH, Bamne M, et al. Joint effect of exposure to infectious agent and host genetic variation on prefrontal structure and function: a model of gene–environment interaction in schizophrenia. Biol Psychiatry 2008; 63 (7 Suppl): 39S. 0_i1092240
- Vollmer-Conna U, Piraino BF, Cameron B, et al. Cytokine polymorphisms have a synergistic effect on severity of the acute sickness response to infection. Clin Infect Dis 2008; 47: 1418-1425. 0_i1092242
- Potvin S, Stip E, Sepehry AA, et al. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry 2008; 63: 801-808. 0_i1092244
- O’Donnell MC, Catts SV, Ward PB, et al. Increased production of interleukin-2 (IL-2) but not soluble interleukin-2 receptors (sIL-2R) in unmedicated patients with schizophrenia and schizophreniform disorder. Psychiatry Res 1996; 65: 171-178. 0_i1092246
- Hickie I, Lloyd A, Wakefield D. Is there a post-infectious fatigue syndrome? Aust Fam Physician 1996; 25: 1847-1852. 0_i1092248
- Maes M, Bosmans E, Suy E, et al. Immune disturbances during major depression: upregulated expression of interleukin-2 receptors. Neuropsychobiology 1990; 24: 115-120. 0_pgfId-1101167
- Hickie I, Hickie C, Lloyd A, et al. Impaired in vivo immune responses in patients with melancholia. Br J Psychiatry 1993; 162: 651-657. 0_i1092251
- Kato T, Monji A, Hashioka S, Kanba S. Risperidone significantly inhibits interferon-gamma-induced microglial activation in vitro. Schizophr Res 2007; 92: 108-115. 0_i1092253
- Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32: 42-48. 0_i1092255
- Hou Y, Wu CF, Yang JY, et al. Effects of clozapine, olanzapine and haloperidol on nitric oxide production by lipopolysaccharide-activated N9 cells. Prog Neuropsychopharmacol Biol Psychiatry 2006; 30: 1523-1528. 0_i1092257
- Miyaoka T, Yasukawa R, Yasuda H, et al. Possible antipsychotic effects of minocycline in patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2007; 31: 304-307. 0_i1092259