




The known Cannabidiol is a non-psychoactive component of the cannabis plant that has anticonvulsant effects in patients with Dravet and Lennox–Gastaut syndromes.
The new Children with drug-resistant epilepsy experienced mild to moderate adverse effects during treatment with cannabidiol; sedation and gastrointestinal effects were the most frequent. Two of 40 participants had abnormal liver function test results.
The implications Cannabidiol is safe in the short term treatment of children with severe, drug-resistant epilepsy. Controlled studies of its therapeutic efficacy are required.
As many as one-third of adults and about 10% of children with epilepsy do not respond adequately to anti-epileptic drugs (AEDs); these patients typically have a variety of comorbid conditions and higher mortality than the general population.1-4 Investigation of cannabinoids as AEDs was stagnant for decades, but has revived dramatically in the past few years, in large part because of public demand for access to medicinal cannabis for patients with epilepsy refractory to other treatments. In particular, the potential benefit of medicinal cannabis for children with drug-resistant epilepsy has been highlighted.5
Cannabidiol, the second most abundant cannabinoid in Cannabis sativa, has attracted interest as an anticonvulsant because of its efficacy in animal models of acute seizure and because it lacks the psychotropic effects of tetrahydrocannabinol.6 However, clinical evidence for its efficacy is limited. Earlier studies had methodological inconsistencies and flaws, particularly the use of non-pharmaceutical grade products. Two recent randomised controlled trials — one in 120 children and young adults (2–18 years) with Dravet syndrome and another in 171 patients (2–55 years) with Lennox–Gastaut syndrome — respectively found significant improvements in convulsive seizure frequency and a drop in seizure frequency during treatment with cannabidiol.7,8
In the United States, an expanded access, multicentre program for providing the pharmaceutical grade cannabidiol oral solution Epidiolex (> 98% purity; 100 mg/mL; GW Pharmaceuticals) to patients with drug-resistant epilepsy was initiated following public demand, despite limited published clinical data. The results of an open label cohort trial in which 162 patients were treated for at least 12 weeks indicated that cannabidiol was well tolerated and may reduce seizure frequency.9
Following an agreement between the New South Wales government and GW Pharmaceuticals, access to Epidiolex was provided for 40 children with drug-resistant epilepsy under the NSW Compassionate Access Scheme (CAS). In this article we describe early Australian experience of the safety and tolerability of cannabidiol as adjunct therapy for treating drug-resistant epilepsy in children.
Methods
Study design
Since August 2016, CAS has provided access to cannabidiol for a maximum of 40 eligible participants at any one time under the Authorised Prescriber Scheme of the Therapeutic Goods Administration.10 Paediatric neurologists with appointments at three sites (Sydney Children’s Hospital, Randwick; Children’s Hospital at Westmead; John Hunter Children’s Hospital, Newcastle) were invited to participate. GW Pharmaceuticals provided Epidiolex at no cost. The CAS remains operational, continuing to enrol patients for treatment with Epidiolex.
Study protocol
Patients who met the eligibility criteria (Box 1) were nominated by their treating paediatric neurologist. Patients were triaged according to epilepsy severity, number of hospital admissions, and age (younger patients preferred). Demographic and disease-related data for the first 40 eligible patients were recorded during the baseline visit, and supplemented by medical records data, including information on severity of disease: aetiology, age of onset, intellectual disability, modified Rankin Scale for Neurological Disability score (range: 0, no symptoms, to 6, death11), previous and current AEDs, non-pharmacological treatment for epilepsy, number of intensive care unit admissions for seizures, and hospital admissions and emergency department presentations during the preceding 12 months. Blood parameters (full blood count, biochemistry, liver function tests, coagulation) were also assessed and electrocardiography undertaken at the baseline visit. The total numbers of emergency rescue medication episodes, episodes of status epilepticus, presentations to emergency departments, and hospital admissions were recorded at baseline (4-week historical control) and throughout treatment. All caregivers were provided with written information about the CAS trial and gave written informed consent.
Enrolled participants were prescribed cannabidiol, but continued their usual AED regime. The initial dose was 5 mg/kg/day in two divided doses, and was increased by 5 mg/kg/day at weekly or longer intervals until the target dose of 25 mg/kg/day was reached. The target dose and dose titration could be altered for clinical reasons, as could the doses of the concomitant AEDs. Participants could continue to receive cannabidiol beyond the 12 weeks of the study if clinically indicated.
Patients attended the clinic for monitoring at 4, 8 and 12 weeks. The current doses of cannabidiol and other drugs, adverse events (including notifiable serious adverse events), and physician and caregiver Global Impression of Change assessments12 were recorded. Quantitative assessment of seizure numbers was not possible because of the severity of disease in our cohort (with uncountable seizures an entry criterion), precluding a standard clinical trial. Instead, caregivers were asked to report their impression of any change from baseline seizure frequency for each seizure type. Blood parameters and levels of other AED drugs were also assessed during the monitoring visits.
Data analysis
Data on clinical features were summarised as numbers or medians (with ranges); means (with ranges) were calculated for the number of status epilepticus episodes, use of midazolam as rescue medication, and presentations to emergency departments or hospital admissions during the 4-week historical baseline and the 3-month trial period, and compared in Student paired t tests. P < 0.05 was deemed statistically significant.
Ethics approval
The protocol for this study was approved by the Sydney Children’s Hospital Network Human Research Ethics Committee (reference, LNR/16/SCHN/226).
Results
Demographic characteristics
Twenty-two boys and 18 girls were enrolled; their mean age was 8.4 years (median, 8.5 years; range, 1.6–16.6 years). Their mean age at diagnosis of epilepsy was 2.5 years (median, 1.1 years; range, 0–12 years). Epileptic encephalopathy (not otherwise specified; ten patients) and focal/multifocal epilepsy (ten patients) were the most frequent syndrome diagnoses (Box 2). Intellectual disability, determined by the treating clinician or by formal developmental assessment, was severe in 17, moderate in 10, mild in 11, and absent in two patients. Functional impairment was assessed with a modified Rankin scale; 22 patients were scored 4–5, indicating that most had moderate to severe disability.11
In the 12 months prior to cannabidiol therapy, the mean number of emergency department presentations or admissions to hospital was 0.29 (range, 0–1.25) per patient per month. The mean lifetime number of admissions to the paediatric intensive care unit for status epilepticus or other complications of seizures was 1.1 (range, 0–7) per patient. Patients had trialled a median of nine AEDs (range, 3–14) before starting cannabidiol therapy, and were currently using a median of three concurrent AEDs (range, 1–5), the five most frequent being clobazam (21 patients), valproate (20), levetiracetam (12), topiramate (10), and lamotrigine (10). Twenty-six patients had undergone non-pharmacological treatments for epilepsy, including 19 patients who had been or were currently on a ketogenic diet, four who had had surgical resections, and three who had had corpus callosotomies, as well as two with vagal nerve stimulators. Four patients had previously trialled artisanal cannabinoids for epilepsy; no information was available on their effect.
Withdrawals
By 12 weeks, there had been four withdrawals (duration of treatment, 2 days to 11.1 weeks). Two withdrew because seizure frequency increased (one withdrawing after 2 days), one because of significant somnolence that resulted in poor feeding and respiratory depression, and one because their transaminase levels were elevated. All other participants continued to receive cannabidiol after conclusion of the study.
Adverse events
Thirty-nine patients reported at least one adverse event, but many were deemed unrelated to cannabidiol treatment; those reported by at least ten participants are listed in Box 3. Intercurrent illness was the most common adverse event (24 patients), and was usually mild and unrelated to cannabidiol treatment. Sixteen participants reported increases in seizure frequency or duration, or episodes of status epilepticus; in two cases, clinicians regarded a relationship with cannabidiol treatment as plausible. Gastrointestinal symptoms (diarrhoea, vomiting, anorexia) were each experienced by seven to nine patients, but were generally mild; diarrhoea and anorexia were attributed to cannabidiol therapy in only four cases.
Fifteen patients reported sedation after a median two weeks’ treatment with cannabidiol (range, one day to 11.5 weeks) and a median dose of 15 mg/kg/day (range, 5–25 mg/kg/day). Ten participants recovered without intervention; the cannabidiol dose was reduced or titration interrupted for four, and for one patient the intervention was unknown. Seven of these 15 patients were also taking clobazam (median dose, 0.86 mg/kg/day; range, 0.40–1.56 mg/kg/day); the median clobazam dose for 14 patients who did not experience sedation was lower (0.44 mg/kg/day; range, 0.09–2.6 mg/kg/day). Blood levels of clobazam and N-desmethylclobazam (norclobazam) could not be assessed.
Two patients had elevated transaminase levels; one withdrew from the trial and the other required temporary interruption of cannabidiol and valproate therapies. Each was receiving 28–35 mg/kg/day valproate.
Serious adverse events
There were 23 serious adverse events in 15 patients that resulted in hospital admission or prolongation of admission, or were deemed important medical events, of which eight events in six patients were considered treatment-related by the treating physician. The most frequent serious adverse events were increased seizure number in eight patients (treatment-related in one patient) and intercurrent illness in five (none treatment-related). Further treatment-related serious adverse events included liver function derangement (resulting in withdrawal) and hyperlipidaemia, each in one patient. One patient had severe somnolence with anorexia and respiratory depression after two weeks of treatment, resulting in prolongation of hospital admission and eventual withdrawal. Supra-therapeutic phenytoin levels with signs of clinical toxicity in two participants were considered to be related to cannabidiol treatment by both treating physicians, and occurred at cannabidiol doses of 10 mg/kg/day (at 4 weeks) and 25 mg/kg/day (at 11.7 weeks) The phenytoin level of one participant had been 19 mg/mL (at the upper end of the therapeutic range, 10–20 mg/mL) prior to commencing cannabidiol therapy.
Other outcomes
Throughout the 12 weeks, 17 patients were admitted to hospital or presented to the emergency department (mean, 0.39 admissions/patient/month; range, 0–2), not significantly different from the rate for the preceding 12 months (P = 0.28). There was no significant change in the number of rescue medication episodes (4-week historical baseline: mean, 0.52 uses/patient/month; range, 0–11; treatment period: mean, 0.58 midazolam uses/patient/month; range, 0–3.7; P = 0.88) or episodes of status epilepticus (4-week historical baseline: mean, 0.25 episodes/patient/month; range, 0–4; treatment period: mean, 0.27 episodes/patient/month; range, 0–2; P = 0.91). No participants reported complete freedom from seizures.
Subgroup analyses
Twelve children were rated much improved or very much improved by their caregiver in Global Impression of Change assessment at 12 weeks; their mean age was 6.3 years (range, 1.9–15.4 years), younger than the other 28 participants (mean, 9.2 years; range, 1.6–15.6 years). The most common syndrome diagnoses in this group were multifocal/focal epilepsy or Dravet syndrome (four children each). The most frequent concomitant AEDs were clobazam (nine children), valproate (seven) and oxcarbazepine (three); 12 of the other 28 children were taking clobazam. According to physician Global Impression of Change assessments, seven participants were much or very much improved, and five were slightly improved (Box 4).
Six children had diagnoses of Dravet syndrome. As rated by their caregivers, one was very much improved, three improved, one slightly improved, and one unchanged after 12 weeks’ cannabidiol treatment; physicians rated none as being very much improved, two as improved, three as slightly improved and one unchanged. All children with Dravet syndrome were taking clobazam (median dose, 0.35 mg/kg/day; range, 0.19–0.71 mg/kg/day).
Discussion
Of 40 children with severe refractory epilepsy who commenced cannabidiol treatment, 36 completed 12 weeks’ therapy with only mild to moderate treatment-related adverse effects, despite multiple comorbid conditions and polypharmacy. Most of the reported adverse events were unrelated to cannabidiol treatment or temporary. The withdrawal rate of 10% was comparable with those of previous trials (13–14%).7,8 Somnolence was the most frequent adverse event that could plausibly be attributed to cannabidiol; it was experienced by nearly 40% of participants, comparable with rates of 14–36% reported by other investigators.7-9 Cannabidiol inhibits the catabolism of an active clobazam metabolite, N-desmethylclobazam, and this may explain why patients taking both drugs are more likely to experience sedation.13 The participants in our cohort who reported somnolence were taking a higher median dose of clobazam than those who did not. Elevated transaminase levels have previously been reported for 1–23% of children receiving cannabidiol, particularly in children taking valproate;7-9,13 our rate of 5% is consistent with these reports. It is important that the liver function of any patient commencing cannabidiol treatment be monitored at regular intervals, especially if they are also taking valproate.
Our study had no objective outcome measure of efficacy, as the strict eligibility criteria (including uncountable seizures) for participation meant that changes in seizure frequency could not be quantified. The mean number of emergency department presentations and hospital admissions increased while taking cannabidiol, as did the numbers of episodes of status epilepticus and the use of rescue medications, but the differences were not statistically significant. Twelve of 36 caregivers reported substantial improvement of their child’s condition in their Global Impression of Change assessments of overall improvement in health,11 as did the clinicians for seven of 37 children. Of the participants rated by caregivers as much or very much improved, nine were taking clobazam, compared with 12 of the other 28 children.
In the expanded access program in the United States, multiple logistic regression analysis found that clobazam use was the only independent factor that predicted a greater than 50% reduction in motor seizure frequency during cannabidiol treatment.9 In the recent Cannabidiol in Dravet Syndrome Study randomised controlled trial, the reduction in median monthly convulsive seizure frequency was significantly greater in the cannabidiol-treated group than in the placebo group (38.9% v 13.3%), achieving a median 22.8% difference (P = 0.01) in convulsive seizure frequency between the two groups.7 As 65% of the participants in the trial were taking clobazam, it has been conjectured that some of the anticonvulsant effect of cannabidiol is related to increased levels of long-acting clobazam metabolites.7,9,14 In the Lennox–Gastaut syndrome study (GWPCARE4), reducing the clobazam dose was necessary for 27% of patients taking cannabidiol.8 It has recently been reported that topiramate and rufinamide levels are also increased during co-administration of cannabidiol.13 As drug interactions are still being elucidated, drug levels of concomitant AEDs and clinical responses should be carefully monitored when initiating cannabidiol treatment.
In our study, caregivers frequently commented on small improvements in cognitive function and in the interaction of their children with their environment; this was not formally assessed and contrasts with almost 40% of participants reporting somnolence. In two small observational studies, improved behaviour or alertness in 25–39%, improved language and motor skills in 8–11%, and improved sleep in 7% of children taking artisanal cannabis extracts were reported by parents.15,16 However, these effects would be difficult to measure objectively; further, a placebo effect is often observed when parents are asked to grade efficacy, as perhaps indicated by the different subjective assessments by physicians and caregivers in our study. This interpretation is supported by the report that patients who moved to Colorado to access medicinal cannabis were more likely to report a benefit than those who already lived there.16
Limitations
The major limitation was the open label design and lack of objective endpoints for assessing participants selected for compassionate access because of the severity of their disease; subjective assessment is liable to bias. Participants who reported a benefit could continue receiving cannabidiol, and this may have influenced self-reported improvement. Additionally, the cohort included children with a heterogeneous group of aetiologies. Finally, the treating clinician could change the doses of cannabidiol and other concurrent AEDs throughout the trial; it is therefore not possible to attribute any benefit to cannabidiol alone.
Conclusion
Drug-resistant epilepsy is an area of significant unmet clinical need, and patients and families are desperate for new treatment options. Randomised controlled trials can help control for the biases that affected our study, and provide data for informing further medical decisions about the patient population suitable for cannabidiol therapy, safety monitoring, and efficacy.
Box 1 – Eligibility and exclusion criteria for participation in trial of cannabidiol for treating drug-resistant epilepsy in children
|
|
|||||||||||||||
|
Eligibility criteria |
|||||||||||||||
|
|
|||||||||||||||
|
Exclusion criteria |
|||||||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
Box 2 – Demographic characteristics at baseline and diagnoses for the 40 participants
|
Characteristic |
|
||||||||||||||
|
|
|||||||||||||||
|
Age (years), median (range) |
8.5 (1.6–16.6) |
||||||||||||||
|
Sex (boys) |
22 |
||||||||||||||
|
Previously trialled medications, median (range) |
9 (3–14) |
||||||||||||||
|
Syndrome |
|
||||||||||||||
|
Focal/multifocal epilepsy |
10 |
||||||||||||||
|
Epileptic encephalopathy (not otherwise specified) |
10 |
||||||||||||||
|
Lennox–Gastaut syndrome |
8 |
||||||||||||||
|
Dravet syndrome |
6 |
||||||||||||||
|
Doose syndrome |
4 |
||||||||||||||
|
Generalised epilepsy |
2 |
||||||||||||||
|
Aetiology |
|
||||||||||||||
|
Genetic |
|
||||||||||||||
|
SCN1A mutation |
6 |
||||||||||||||
|
MECP2 mutation |
2 |
||||||||||||||
|
Other confirmed genetic basis |
1 |
||||||||||||||
|
Presumed genetic basis |
19 |
||||||||||||||
|
Structural |
|
||||||||||||||
|
Malformations of cortical development |
6 |
||||||||||||||
|
Neurodegenerative |
1 |
||||||||||||||
|
Post-encephalitic epilepsy |
1 |
||||||||||||||
|
Hypoxic–ischaemic encephalopathy |
1 |
||||||||||||||
|
Tuberous sclerosis |
2 |
||||||||||||||
|
Other neurocutaneous type |
1 |
||||||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
Box 3 – Adverse events most frequently reported by 40 children during first 12 weeks of treatment with cannabidiol for drug-resistant epilepsy
|
Adverse event |
Number of participants with event |
|
Cannabidiol therapy: median (range) |
||||||||||||
|
Total |
Cannabidiol therapy-related |
Median severity of event |
Dose (mg/kg/day) |
Duration (weeks) |
|||||||||||
|
|
|||||||||||||||
|
Benign viral illness |
24 |
0 |
Mild |
20 (5–25) |
3.1 (0.1–11) |
||||||||||
|
Increased seizures |
16 |
2 |
Moderate |
25 (5–25) |
7.8 (0.3–15) |
||||||||||
|
Somnolence |
15 |
13 |
Mild |
15 (5–25) |
2.0 (0.1–11) |
||||||||||
|
Diarrhoea |
9 |
4 |
Mild |
20 (5–25) |
3.4 (0.1–7.9) |
||||||||||
|
Anorexia |
7 |
4 |
Mild |
15 (5–25) |
2.0 (0.1–8.9) |
||||||||||
|
Vomiting |
7 |
1 |
Mild |
25 (5–25) |
5.7 (0.1–11) |
||||||||||
|
Rash |
4 |
1 |
Mild |
15 (5–25) |
2.3 (0.7–5.9) |
||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
Box 4 – Caregiver and physician Global Impression of Change assessments for 40 children with drug-resistant epilepsy after 12 weeks’ treatment with cannabidiol*
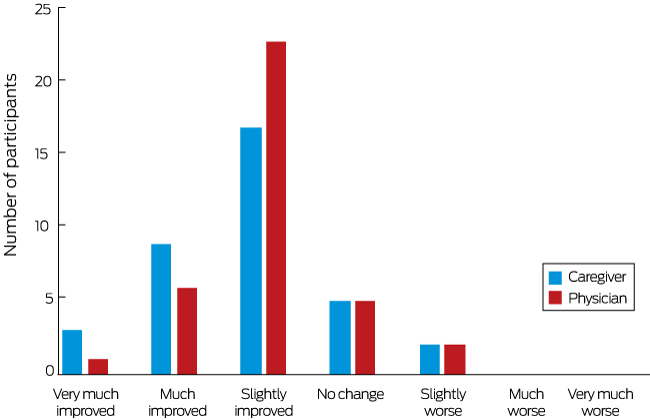
* Responses for the patient who was withdrawn at 11.4 weeks are included, but not those for the three patients who withdrew earlier; one further caregiver did not provide a rating.
Received 7 January 2018, accepted 4 May 2018
- Kerrie-Anne Chen1
- Michelle Farrar1,2
- Michael Cardamone1,2
- Deepak Gill3
- Robert Smith4,5
- Christopher T Cowell6
- Linda Truong7
- John A Lawson1,2
- 1 Sydney Children's Hospital Randwick, Sydney, NSW
- 2 UNSW Sydney, Sydney, NSW
- 3 Children's Hospital at Westmead, Sydney, NSW
- 4 John Hunter Children's Hospital, Newcastle, NSW
- 5 University of Newcastle, Newcastle, NSW
- 6 Kids Research Institute, Sydney Children's Hospitals Network, Sydney, NSW
- 7 Clinical Trials Unit, Sydney Children's Hospitals Network, Sydney, NSW
The New South Wales Compassionate Access Scheme for Refractory Epilepsy was funded by the NSW Government. GW Pharmaceuticals supplied Epidiolex on a compassionate access basis. We acknowledge the support of the NSW Ministry of Health, and the participation of Kerry Chant and Jan Fizzell; the original project manager Michael Revius; the treating clinicians Ian Andrews, Jayne Antony, Simone Arden-Holmes, Annie Bye, Russell Dale, Sachin Gupta, Alexandra Johnson, Manoj Menezes, Christina Miteff, Sekhar Pillai, Peter Procopis, Hugo Sampaio, Gopinath Subramanian, Christopher Troedson, Richard Webster, Helen Young; and the research nurses Stephanie Richardson, Naomi Adams, Aimee Williams, Samantha Woods, Elaine Coleman, Victoria Middleton, Katharine McCreath, Janis Brown, Regienald Gayaman, Pamela Cheung, Jennifer Lampard and Elizabeth Clarke.
John Lawson is the lead investigator in the NSW Ministry of Health-funded medical cannabis trials. Deepak Gill and Robert Smith are sub-investigators on the NSW Ministry of Health-funded medical cannabis trials.
- 1. World Health Organization, International League Against Epilepsy, International Bureau for Epilepsy. Atlas: epilepsy care in the world. Geneva: WHO, 2005 http://www.who.int/mental_health/publications/atlas_epilepsy_care_2005/en/ (viewed June 2018).
- 2. Geerts A, Arts WF, Stroink H, et al. Course and outcome of childhood epilepsy: a 15-year follow-up of the Dutch Study of Epilepsy in Childhood. Epilepsia 2010; 51: 1189-1197.
- 3. Berg AT, Shinnar S, Levy SR, et al. Early development of intractable epilepsy in children: a prospective study. Neurology 2001; 56: 1445-1452.
- 4. Chen KA, Farrar MA, Cardamone M, et al. Cannabis for paediatric epilepsy: challenges and conundrums. Med J Aust 2018; 208: 132-136. <MJA full text>
- 5. Young S. Marijuana stops child’s severe seizures [internet]. CNN 7 Aug 2013. http://edition.cnn.com/2013/08/07/health/charlotte-child-medical-marijuana/ (viewed June 2018).
- 6. Longo DL, Friedman D, Devinsky O. Cannabinoids in the treatment of epilepsy. N Engl J Med 2015; 373: 1048-1058.
- 7. Devinsky O, Cross JH, Laux L, et al; Cannabidiol in Dravet Syndrome Study Group. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med 2017; 376: 2011-2020.
- 8. Thiele EA, Marsh ED, French JA, et al; GWPCARE4 Study Group. Cannabidiol in patients with seizures associated with Lennox–Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018; 391: 1085-1096.
- 9. Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol 2016; 15: 270-278.
- 10. Australian Government Department of Health, Therapeutic Goods Administration. Authorised Prescriber Scheme. 3 July 2017. https://www.tga.gov.au/authorised-prescriber-scheme (viewed July 2017).
- 11. Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials. A literature review and synthesis. Stroke 2007; 38: 1091-1096.
- 12. Busner J, Targum SD. The Clinical Global Impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont) 2007; 4: 28-37.
- 13. Geffrey AL, Pollack SF, Bruno PL, et al. Drug–drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015; 56: 1246-1251.
- 14. Tang R, Fang F. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med 2017; 377: 699.
- 15. Treat L, Chapman KE, Colborn KL, et al. Duration of use of oral cannabis extract in a cohort of pediatric epilepsy patients. Epilepsia 2017; 58: 123-127.
- 16. Press CA, Knupp KG, Chapman KE. Parental reporting of response to oral cannabis extracts for treatment of refractory epilepsy. Epilepsy Behav 2015; 45: 49-52.
Abstract
Objective: To evaluate the tolerability and safety of cannabidiol for treating drug-resistant epilepsy in children, and to describe adverse events associated with such treatment.
Study design: Prospective, open label cohort study.
Setting: Three tertiary NSW referral centres with paediatric neurology services.
Participants: First 40 children enrolled in the NSW Compassionate Access Scheme for children with drug-resistant epilepsy and uncountable daily seizures.
Intervention: Children received cannabidiol as an adjunct anti-epileptic drug, titrated to a maximum of 25 mg/kg/day, for up to 12 weeks.
Outcome measures: Adverse events, withdrawals, and caregiver and physician Global Impression of Change assessments were recorded at 4, 8 and 12 weeks. Seizure frequency could not be reliably recorded because of disease severity.
Results: Thirty-nine patients reported at least one adverse event; many were deemed unrelated to cannabidiol treatment. The most frequent treatment-related adverse event was somnolence (15 participants), which resolved spontaneously in ten patients; it was particularly frequent in patients taking higher clobazam doses. Gastrointestinal effects (nausea, vomiting, diarrhoea) were each reported by seven to nine participants. Four children were withdrawn from treatment, including one with elevated transaminase levels. The caregivers of 12 children felt the overall health of their children had much or very much improved; clinicians assessed seven children as being much or very much improved.
Conclusion: Cannabidiol as an adjunct treatment had some subjective benefit for overall health, with a manageable adverse event profile. Monitoring changes in liver function and awareness of potential drug interactions is essential. Whether the reported benefit is attributable to cannabidiol cannot be established in an open label study of participants with severe intractable epilepsy.