





Volume 206 - Issue 8
Motor neurone disease: progress and challenges
Authors: Thanuja Dharmadasa, Robert D Henderson, Paul S Talman, Richard AL Macdonell, Susan Mathers, David W Schultz, Merrillee Needham, Margaret Zoing, Steve Vucic and Matthew C Kiernan
Med J Aust 2017; 206 (8): 357-362. || doi: 10.5694/mja16.01063
Published online: 1 May 2017
Published online: 1 May 2017
Recent evolution in the clinical, pathological and genetic understanding of MND is progressively unmasking the multifactorial nature of this complex condition
Summary
- Major progress has been made over the past decade in the understanding of motor neurone disease (MND), changing the landscape of this complex disease.
- Through identifying positive prognostic factors, new evidence-based standards of care have been established that improve patient survival, reduce burden of disease for patients and their carers, and enhance quality of life. These factors include early management of respiratory dysfunction with non-invasive ventilation, maintenance of weight and nutritional status, as well as instigation of a multidisciplinary team including neurologists, general practitioners and allied health professionals.
- Advances in technology have enhanced our understanding of the genetic architecture of MND considerably, with implications for patients, their families and clinicians. Recognition of extra-motor involvement, particularly cognitive dysfunction, has identified a spectrum of disease from MND through to frontotemporal dementia.
- Although riluzole remains the only disease-modifying medication available in clinical practice in Australia, several new therapies are undergoing clinical trials nationally and globally, representing a shift in treatment paradigms. Successful translation of this clinical research through growth in community funding, awareness and national MND research organisations has laid the foundation for closing the research–practice gap on this debilitating disease.
- In this review, we highlight these recent developments, which have transformed treatment, augmented novel therapeutic platforms, and established a nexus between research and the MND community. This era of change is of significant relevance to both specialists and general practitioners who remain integral to the care of patients with MND.
Motor neurone disease (MND) is a progressive, neurodegenerative disorder that mainly attacks the human motor system, leading to significant disability and ultimately death, usually within 3 years.1 The incidence of MND in Western populations, including Australia, is about 2–3 per 100 000, with a national prevalence of about 8 per 100 000.2 Currently, 1500 Australian patients suffer from the disease.3 As there remains no test for MND, diagnosis is based on clinical findings, supported by investigations such as neurophysiological testing and structural imaging to exclude mimic disorders.4 Given that MND is clinically and pathologically heterogeneous, therapeutic and neuroprotective targets have been difficult to identify. However, progress in recent years has stimulated innovative research into this devastating disorder. In this review, we discuss areas of progress in the field of MND, including improved understanding of the various clinical phenotypes, the development of standards of care, the continuum with frontotemporal dementia (FTD), the role of genetics, and the global clinical trials pipeline. We also highlight the importance of translating research into clinical practice through various networks.
To formulate an evidence-based review of MND in clinical practice, we searched PubMed for original and review articles published between 1990 and 2016, focusing on publications within the past 5 years. We also sourced guidelines and other articles from MND Australia and the Cochrane Database of Systematic Reviews, and drew on collective specialist experience across Australia’s multidisciplinary clinics.
New clinical perspectives
Patients with MND are heterogeneous with varied presentations, depending on the site of disease onset. The definitive clinical characteristics remain the presence of upper and lower motor neurone signs coexisting in the same symptomatic area (Box 1).3,5 The median time to diagnosis is about 14 months, which is often a distressing period that also inevitably delays appropriate disease-modifying therapies and acceptance of prognosis.4,6
The clinical phenotype and site of disease onset appears to be of important prognostic significance in MND.7 Four main clinical phenotypes are described based on the relative degree of upper and lower motor neurone predominance and the site of onset:
Amyotrophic lateral sclerosis (ALS) represents 70% of cases.3,6 ALS classically begins in the limb and exhibits a combination of upper and lower motor neurone signs.3,6 In about 20% of patients, the weakness starts in the bulbar region.3 With a median survival of about 3 years, ALS is the most malignant of the MND phenotypes.7
Isolated bulbar palsy represents about 4–8% of cases.8 Patients have localised, progressive speech and swallowing difficulties for a prolonged period (> 6 months), despite relative preservation of limb strength. Although almost all isolated bulbar palsy patients eventually progress to definite ALS, they have a better prognosis than bulbar-onset ALS, with disease duration extended by at least 12 months.8
Progressive muscular atrophy presents with pure lower motor neurone signs and represents 5% of the MND phenotype.9 A subgroup develops a flail limb variant, in which symptoms are limited either to the upper (flail arm syndrome) or the lower limbs (flail leg syndrome) for at least 12 months. The prognosis for these variants is typically more prolonged than for classic ALS, but patients with more generalised weakness (> 50% of limb regions affected) follow a similar disease course to ALS.9
Primary lateral sclerosis (1–3% of cases)10 presents with pure upper motor neurone signs and a predilection for lower limb disease onset. Patients experience a much slower disease progression and better prognosis, with some known to have normal life expectancy.10
Advances in treatment approach: new standards of care
An increased understanding of this complex disorder has enabled identification of several important factors that improve survival and reduce patient symptoms, generating new evidence-based treatment interventions.11
Multidisciplinary care
Multidisciplinary care has been reported to improve both quality of life and survival, potentially up to 7–24 months.12,13 For patients with bulbar disease, survival benefit is possibly even longer.12 The care team involves medical, nursing and allied health professionals, and focuses on proactive intervention for early holistic management of the patient (Box 2). The network also includes input from MND Australia and state-based associations, which assist with care coordination, family support and community education. This dynamic framework addresses the complex medical issues that arise over time, allows for continual assessment of functional disabilities and psychosocial burden, and provides a network of support for the treating clinicians.13 Management within a multidisciplinary care clinic is therefore recommended for all patients, with such clinics operating in most capital cities around Australia.2 Telemedicine services are also available in some areas for patients who have attendance difficulties (such as due to disability or location).
Respiratory management with non-invasive ventilation
The benefit of using non-invasive positive pressure ventilation for respiratory involvement has been a crucial discovery in MND care, not only because it greatly improves symptoms and quality of life, but because it extends patient survival by up to 13 months.14,15 Only a small proportion of patients with MND have respiratory problems at the initial onset of disease, but almost all will develop symptoms during the course of the disease and most will die from respiratory complications.16,17 Respiratory symptoms should therefore be assessed at each visit and, if tolerated, early institution of non-invasive positive pressure ventilation should be implemented.
Nutritional support
Weight loss and malnutrition during the course of disease exert a negative effect on survival, associated with more rapid disease progression.18 This can occur from development of a hypermetabolic state, swallowing problems, neuropsychiatric issues, and feeding difficulties due to loss of limb function.19 Monitoring patients for weight loss is thus essential, and managing this with the help of an experienced speech pathologist and dietitian is critical in routine MND care. Interventions for enteral feeding options (such as percutaneous endoscopic gastrostomy tubes) are helpful in circumventing dysphagia (especially in patients with bulbar-onset MND) and for maintaining nutrition in patients using long term ventilatory support.20,21
Other symptoms
Cumulative experience has guided current clinical practice for the treatment of other symptoms often experienced by patients with MND related to their progressive motor and non-motor dysfunction (Box 3).
End-of-life challenges
For all patients with MND, the issue of advanced care planning needs to be raised within an appropriate timeframe by the multidisciplinary care team. Although the end-of-life phase remains poorly defined and timing of palliative care input is not consistent globally,22 advance care planning assists patients and their families with important decision making, imparting control and composure in a situation in which they may otherwise be absent. Carers have identified disease-specific advanced directives (such as patient letters of future care) as useful tools to stimulate such discussion while maintaining respect for patient autonomy.23 Appropriate timing for these discussions is often individually based, but can be sensitively approached when introducing intervention options such as non-invasive positive pressure ventilation or gastrostomy.
The MND–FTD spectrum
Although originally thought to be a purely motor disorder, MND has been increasingly recognised for its extra-motor manifestations. Cognitive impairment is common and may develop in up to 50% of patients, manifesting as language abnormalities and mild to moderate frontal dysfunction.24 The presence of cognitive impairment is associated with a negative impact on survival, more rapid disease progression, decreased functional ability, higher rates of non-compliance with therapy, and increased psychosocial distress and burden for carers.24
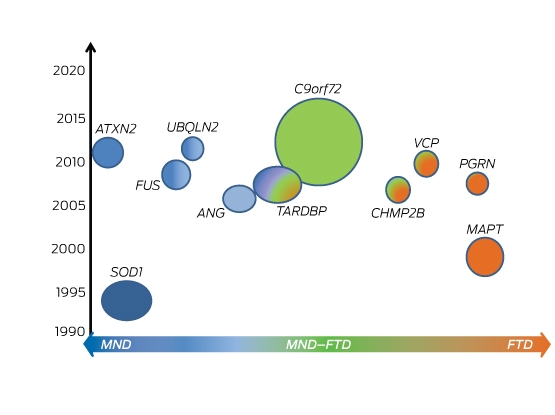
About 15% of patients with MND who have cognitive impairment meet the criteria for FTD, and are considered to have MND–FTD.25 MND and FTD are known to share distinct clinical, neuropathological and genetic features, and it is now recognised that they are part of a continuum in which pure MND (with no cognitive involvement) and pure FTD (with no motor involvement) form ends of a spectrum.25 In 2011, the discovery of a mutation in the C9orf72 gene unambiguously linked these two conditions (Box 4).26,27 C9orf72 mutation is a hexanucleotide repeat expansion, now known to be responsible for about 40% of familial MND, 25% of familial FTD, and up to 80% of MND–FTD cases.27
Genetics: new insights
The recent discovery of novel genes associated with MND has changed the genetic landscape and encouraged the possibility of future gene therapy. Increasingly, this is modifying the clinical approach to MND, and clinicians commonly face questions regarding genetic causes, testing and family risk. Although mainly driven by the neurologist and clinical geneticist, the dilemma on handling such queries necessitates an understanding of appropriate genetic testing options and counselling for patients and their relatives.
Both familial and sporadic MND are clinically similar, and the genetic and biological distinction between them is also becoming increasingly blurred. Ninety per cent of MND cases appear to be sporadic, with 5–10% of patients having a family history of MND.28 Family history is established with the presence of MND or FTD in a first- or second-degree relative. This is most commonly inherited in an autosomal dominant manner but may be autosomal recessive or X-linked.29 Empirical data suggest that the lifetime risk of MND in first-degree relatives of sporadic patients is 1–3%, with a lower risk for second-degree relatives and no apparent increase in risk for more distant relations.30 However, many factors can complicate the pattern and presence of inheritance, including incomplete family information, false paternity, early death and non-penetrance.
To date, more than 25 MND genes have been discovered, explaining 10% of sporadic and 65% of familial disease.31 The C9orf72 repeat expansion is the most common genetic cause of MND. This is reported to account for about 40% of familial and about 7% of sporadic disease,31,32 but the exact frequency of MND-related genes varies between different populations and specific data on prevalence in Australia are still limited. Preliminary national studies identified C9ORF72 mutation in 38.5% of familial cases and 3.5% of sporadic cases.33 There are also some shared clinical traits in these patients, who are typically of northern European descent and who clinically often have neuropsychiatric symptoms, including frank FTD.32,33 There is also a suggestion of higher frequency of bulbar-onset disease, earlier age of symptom onset, and more rapid disease progression.26,32
Who should be offered testing?
Currently, as there is no proven lifestyle modification or medication to reduce the risk of disease, the reason for genetic testing is mainly to provide diagnostic support or pre-natal counselling.34 The option for testing is usually offered by a neurologist to symptomatic patients who have a first- or second-degree relative with MND, FTD or MND–FTD. It can also be discussed with all other symptomatic patients, but with emphasis on the uncertainties of testing. Guidelines do not recommend that asymptomatic at-risk people be routinely offered testing, particularly as positive test results do not reliably correlate with disease development, severity, progression or age of disease onset.34
Genetic testing options
Until recently, testing was limited to a single gene — SOD1. Multigene next-generation sequencing panels and whole-exome sequencing in particular have significantly increased the identification of new genes and commercial testing options. Most tests take from 3–24 weeks for results. Although some genetic services may provide subsidised testing for several MND-related genes, there is no Medicare rebate for testing. The test can therefore cost between $250 and $9000, depending on the test and the laboratory.25,33
Limitations of testing
Despite the increase in genetic understanding of disease, establishing genetic testing guidelines has been complicated because of areas of persisting uncertainty. Establishing whether a mutation is pathogenic can be difficult, even for widely studied genes.34 There are also high rates of variants of uncertain significance in multigene panels, and technical challenges from differing laboratory techniques also generate problems.35 These limitations in testing should be emphasised to the patient, including the fact that positive results do not predict disease course, negative results do not exclude genetic basis of disease, and results may not provide interpretable information if a genetic variant of uncertain significance is identified.
Genetic counselling
Given the complications, interpreting results correctly and counselling patients with accurate risk assessments can be difficult. Genetic counselling should be managed by the multidisciplinary team, mainly via a clinical geneticist, but also involving a neurologist, general practitioner and psychologist. Genetic counselling clinics operate across Australia (information is available at http://www.genetics.edu.au). Patients and family members should have pre-test consultation with a genetics counsellor as a prerequisite before undergoing a test. Post-test counselling is usually offered for patients with a positive test result, and implications for family members (including offspring) can be discussed. DNA banking permits future testing and is an option for families who do not feel ready to undergo genetic testing.32,34
Neuroprotection: current and future
Riluzole currently remains the only neuroprotective medication available for patients with MND, and early commencement is recommended for all patients.36,37 Riluzole modulates sodium channels and inhibits glutamate release, providing a survival benefit of about 3–6 months, potentially greater for patients with bulbar-onset disease.38 It has a modest side effect profile and is generally well tolerated by patients. Liver function tests and a full blood count should be carried out monthly for 3 months, and 3-monthly thereafter, with treatment cessation if liver function test results (alanine transaminase and aspartate transaminase levels) exceed more than five times the upper limit of normal and/or neutropenia develops.37,39
Current trials
Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one) is a free radical scavenger that has gained attention as a potential agent to slow disease progression in MND. Originally approved in Japan in 2002 to treat ischaemic stroke, edaravone has been shown to have multiple effects on the neural and vascular ischaemic cascade.40 Although an initial phase 3 randomised controlled trial in MND found no clinically significant effect, a subgroup of mildly symptomatic MND patients who had a forced vital capacity ≥ 80% and who were not more than 2 years from symptom onset showed slowing of disease progression.41 Intravenous edaravone treatment for such patients was approved in Japan in 2015. An oral equivalent is currently being tested in Europe in a phase 2 trial (http://www.treeway.nl/news-treeway-announces-positive-data-phase-1-trial-tw001).
There have been several other phase 3 randomised control trials that have shown promising results. A 2015 trial of ultra-high dose methylcobalamin showed a dose-dependent prolongation of survival when used early in disease.42 Positive results have also been seen with masitinib, an oral tyrosine kinase inhibitor that targets macrophages and mast cells,43 and a current trial has shown improvement in patients’ Revised ALS Functional Rating Scale scores and forced vital capacity (https://www.clinicaltrialsregister.eu/ctr-search/trial/2010-024423-24/IE#E). Although not available for use in Australia, masitinib has recently been granted orphan drug status by the European Medicines Agency and by the United States Food and Drug Administration (https://alsnewstoday.com/news-posts/2016/08/11/ab-science-potential-als-treatment-masitinib-named-orphan-drug-by-ema).
In Australia, three human clinical trials are currently commencing in Sydney and Melbourne. The first will analyse central nervous system copper treatment in patients with MND, after delivery of a copper compound (CuATSM) into the central nervous system of SOD1(G93A) mouse models prolonged survival by 18 months.44 The second trial is a phase 2 study assessing the effects of the antiretroviral agent Triumeq (ViiV Healthcare; combination tablet containing 600 mg abacavir, 300 mg lamivudine and 50 mg dolutegravir) in treating MND. The basis for this trial relates to links between human endogenous retrovirus K and the development of MND in mouse models and in humans.45 The third trial is a randomised crossover study evaluating the efficacy of oral FLX-787 (Flex Pharma) — a constituent of ginger — for patients with MND suffering from muscle cramps and spasticity. This study hypothesises that FLX-787 activates transient receptor potential ion channels involved in pain, and indirectly decreases motor neurone hyperexcitability.46 Recruitment for these trials began at involved centres towards the end of 2016.
Gene therapy
Gene technology aims to protect motor neurones by modulating mutated gene expression and reducing toxic RNA and proteins. Encouragingly, studies in SOD1 mouse models have shown significant benefits,47 with C9orf72 studies ongoing. However, this area is still in its infancy, mainly due to the complexity of the MND genetic environment, the difficulty in correlating mutations with clinical pathogenicity, the technical difficulty of the treatment itself, and the economic challenge it presents.47
Stem cell therapy
The benefit of stem cell therapy is frequently questioned by patients and families. The efficacy of this treatment remains open in the literature, with small phase 2 trials from around the world reporting safety and efficacy using various modes of stem cell delivery.48 Human induced pluripotent stem cell technology is a new and evolving field in which cells are created from patients with MND and differentiated into relevant cell subtypes, such as motor neurones and astrocytes. This has been used in vitro and in mouse models, but engraftment into patients with MND for therapeutic benefit remains a challenge.49 Although some countries offer therapeutic trials of stem cell treatment, it is not offered in Australia owing to its unclear efficacy. More understanding of underlying mechanistic processes and long term safety data are needed before clinical translation will be possible.
Fundraising, awareness and research
While the search for improved therapies continues, strong patient advocacy and home-based assistance has been provided by state-based MND associations and MND Australia. The Australian Motor Neurone Disease Registry also collects information from MND centres across Australia to increase clinical understanding and improve quality of care.2
Support for trials in Australia is also needed to drive national research and provide Australian patients with access to potential international treatments. Recently, Australian Clinical Trials and Translational Research Advisory (https://curemnd.org.au/meet-the-team) has been established to enable collaboration and grant this much needed opportunity to patients. Increase in community awareness (through campaigns such as the “ice bucket challenge”) and the generation of funding in Australia via organisations such as Cure for MND Foundation (https://www.curemnd.org.au) and the MND Research Institute of Australia (MNDRIA) (http://www.mndaust.asn.au) has supported MND national research in an unprecedented way, and is enabling successful translation of research by clinicians into the MND community.
Conclusion
Recent evolution in the clinical, pathological and genetic understanding of MND is progressively unmasking the multifactorial nature of this complex condition. Up-to-date knowledge of the current climate is thus essential for optimal patient care. Closing the research–practice gap through growth of community awareness and support from MND organisations has also been critical for this process. Ongoing engagement of professionals and the community is an invaluable asset to patients, encouraging novel therapeutic strategies and powering the drive to find effective treatments.
Box 1 – Clinical signs and symptoms of upper and lower motor neurone involvement in motor neurone disease
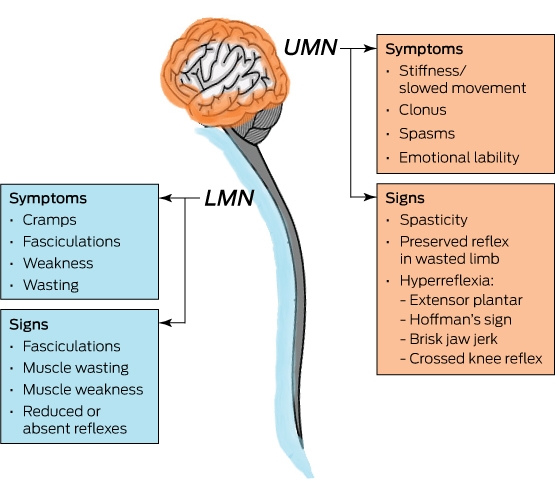
UMN = upper motor neurone. LMN = lower motor neurone.
Box 2 – Motor neurone disease management: multidisciplinary care model
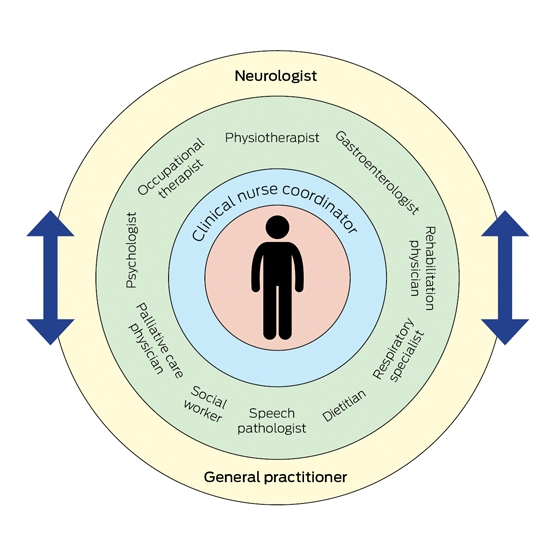
The multidisciplinary care model centres on the patient with motor neurone disease. It involves dynamic integration of medical, nursing and allied health professionals for optimal patient management. Care is often coordinated by the clinical nurse, with the neurologist and general practitioner overseeing all aspects of care.
Box 3 – Symptomatic treatments for motor neurone disease
Symptom |
Pharmacological management (first-line medications) |
Non-pharmacological management |
|||||||||||||
Secondary to motor dysfunction |
|||||||||||||||
Cramps/fasciculations |
Magnesium; carbamazepine |
Physiotherapy (stretches); massage; hydrotherapy |
|||||||||||||
Spasticity |
Baclofen; clonazepam |
Physiotherapy; hydrotherapy |
|||||||||||||
Dyspnoea |
Morphine (oral)*; lorazepam |
Ventilatory support (respiratory review); chest physiotherapy; manually assisted coughing |
|||||||||||||
Thickened saliva |
Normal saline nebulisers; nebulised mucolytics (eg, N-acetylcysteine) |
Natural mucolytics (papaya, pineapple, dark grape juice); hydration; portable suction device |
|||||||||||||
Excess (watery) saliva |
Amitriptyline; atropine (sublingual)*; glycopyrrolate |
Portable suction device; diligent mouth care |
|||||||||||||
Laryngospasm/paroxysmal choking |
Lorazepam (sublingual)*; morphine (oral)* |
Careful positioning; suctioning; ± ventilatory support |
|||||||||||||
Secondary to non-motor dysfunction |
|||||||||||||||
Pain† |
Musculoskeletal: paracetamol; ibuprofenNeuropathic: gabapentin, pregabalin |
Physiotherapy; hydrotherapy; pressure area care (repositioning, pressure cushion/mattress) |
|||||||||||||
Cognitive dysfunction |
Memantine‡; antidepressants |
Education of caregivers/family |
|||||||||||||
Emotional lability; depression |
Amitriptyline; citalopram; mirtazapine |
Psychological support; cognitive behavioural therapy |
|||||||||||||
Sleep disturbance |
Amitriptyline; benzodiazepines |
Address underlying problem; may need respiratory review ± non-invasive ventilation |
|||||||||||||
Constipation |
Aperients; suppositories |
Dietary changes (increased fibre and fluid); review drug adverse effects |
|||||||||||||
* The specific formulation indicated is preferred for treatment of this symptom. † Pain is often multifactorial, and treatment must therefore be tailored to the individual cause(s). ‡ Used off label, but not supported by a recent negative phase 3 trial in FTD (Boxer AL, Knopman DS, Kaufer DI, et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2013; 12: 149-156). | |||||||||||||||
Competing interests
No relevant disclosures.
Acknowledgements
Thanuja Dharmadasa is a recipient of an Australian Postgraduate Award (University of Sydney), a Rotary Club of Cronulla Funding Partner Scholarship, a MNDRIA PhD Top-up Grant and a Yulgilbar Foundation Alzheimer’s Research Program PhD Top-up Award. Matthew Kiernan is the Editor-in-Chief of the Journal of Neurology, Neurosurgery and Psychiatry. This work was supported by funding to ForeFront, a collaborative research group dedicated to the study of FTD and MND, from a National Health and Medical Research Council of Australia program grant (1037746).
References
- Traxinger K, Kelly C, Johnson BA, et al. Prognosis and epidemiology of amyotrophic lateral sclerosis: analysis of a clinic population, 1997-2011. Neurol Clin Pract 2013; 3: 313-320.
- Kiernan MC, Talman P, Henderson RD, et al. Establishment of an Australian motor neurone disease registry. Med J Aust 2006; 184: 367-368.
- Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011; 377: 942-955.
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 2011; 7: 639-649.
- Hobson E, Harwood C, McDermott CJ, Shaw PJ. Clinical aspects of motor neurone disease. Medicine 2016; 44(9): 552-556.
- Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 2014; 37: 433-442.
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014; 10: 661-670.
- Burrell JR, Vucic S, Kiernan MC. Isolated bulbar phenotype of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2011; 12: 283-289.
- Visser J, van den Berg-Vos RM, Franssen H, et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol 2007; 64: 522-528.
- Gordon PH, Cheng B, Katz IB, et al. The natural history of primary lateral sclerosis. Neurology 2006; 66: 647-653.
- Birks C, Egan M, Rizk R, et al. Motor neurone disease: aspects of care for the primary health care team. Sydney: MND Australia, 2014. http://www.mndcare.net.au/Overview/MNDcare-approach/Information-and-discussion/For-health-and-community-care-professionals/MND-Australia-2014/MND-Aspects-of-care-for-the-primary-health-car-(1).aspx (accessed Dec 2016).
- Rooney J, Byrne S, Heverin M, et al. A multidisciplinary clinic approach improves survival in ALS: a comparative study of ALS in Ireland and Northern Ireland. J Neurol Neurosurg Psychiatry 2015; 86: 496-501.
- Traynor BJ, Alexander M, Corr B, et al. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study. J Neurol Neurosurg Psychiatry 2003; 74: 1258-1261.
- Kleopa KA, Sherman M, Neal B, et al. Bipap improves survival and rate of pulmonary function decline in patients with ALS. J Neurol Sci 1999; 164: 82-88.
- Berlowitz DJ, Howard ME, Fiore JF, et al. Identifying who will benefit from non-invasive ventilation in amyotrophic lateral sclerosis/motor neurone disease in a clinical cohort. J Neurol Neurosurg Psychiatry 2016; 87: 280-286.
- Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009; 73: 1218-1226.
- Corcia P, Pradat P-F, Salachas F, et al. Causes of death in a postmortem series of ALS patients. Amyotroph Lateral Scler 2008; 9: 59-62.
- Desport JC, Preux PM, Truong TC, et al. Nutritional status is a prognostic factor for survival in ALS patients. Neurology 1999; 53: 1059-1063.
- Desport JC, Preux PM, Truong CT, et al. Nutritional assessment and survival in ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 91-96.
- Spataro R, Ficano L, Piccoli F, et al. Percutaneous endoscopic gastrostomy in amyotrophic lateral sclerosis: effect on survival. J Neurol Sci 2011; 304: 44-48.
- Wills A-M, Hubbard J, Macklin EA, et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014; 383: 2065-2072.
- Connolly S, Galvin M, Hardiman O. End of life management in patients with amyotrophic lateral sclerosis. Lancet Neurol 2015; 14: 435-442.
- Murray L, Butow PN, White K, et al. Advance care planning in motor neuron disease: a qualitative study of caregiver perspectives. Palliative Medicine 2016; 30(5): 471-478.
- Phukan J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2012; 83: 102-108.
- Burrell JR, Halliday GM, Kril JJ, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016; pii: S0140-6736(16) 00737-00746.
- Snowden JS, Harris J, Richardson A, et al. Frontotemporal dementia with amyotrophic lateral sclerosis: a clinical comparison of patients with and without repeat expansions in C9orf72. Amyotroph Lateral Scler Frontotemporal Degener 2013; 14: 172-176.
- Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012; 11: 323-330.
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 2011; 7: 603-615.
- Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta 2006; 1762: 956-972.
- Hanby MF, Scott KM, Scotton W, et al. The risk to relatives of patients with sporadic amyotrophic lateral sclerosis. Brain 2011; 134: 3454-3457.
- Rheenen W, Shatunov A, Dekker AM, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 2016; 48: 1043-1048.
- Roggenbuck J, Quick A, Kolb SJ. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians. Genet Med 2017; 19: 267-274.
- Williams KL, Fifita JA, Vucic S, et al. Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry 2013; 84: 931-935.
- Chio A, Battistini S, Calvo A, et al. Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry 2014; 85: 478-485.
- Akimoto C, Volk AE, van Blitterswijk M, et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet 2014; 51: 419-424.
- McDermott CJ, Shaw PJ. Diagnosis and management of motor neurone disease. BMJ 2008; 336: 658-662.
- Zoing MC, Burke D, Pamphlett R, et al. Riluzole therapy for motor neurone disease: an early Australian experience (1996–2002). J Clin Neurosci 2006; 13: 78-83.
- Zoccolella S, Beghi E, Palagano G, et al. Riluzole and amyotrophic lateral sclerosis survival: a population-based study in southern Italy. Eur J Neurol 2007; 14: 262-268.
- Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 2012; (3): CD001447.
- Yoshida H, Yanai H, Namiki Y, et al. Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury. CNS Drug Rev 2006; 12: 9-20.
- Abe K, Itoyama Y, Sobue G, et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener 2014; 15: 610-617.
- Kaji R, Kuzuhara S, Iwasaki Y, et al. Ultra-high dose methylcobalamin (E0302) prolongs survival of ALS: report of 7 years’ randomised double-blind, phase 3 clinical trial (ClinicalTrials.gov NCT00444613). Neurology 2015; 84 (Suppl): 7060.
- Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One 2009; 4: e7258.
- Williams JR, Trias E, Beilby PR, et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the copper-chaperone-for-SOD. Neurobiol Dis 2016; 89: 1-9.
- Li W, Lee M-H, Henderson L, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 2015; 7: 307ra153.
- Meotti FC, Lemos AE, Calixto JB. TRP modulation by natural compounds. Handb Exp Pharmacol 2014; 223: 1177-1238.
- Scarrott JM, Herranz-Martín S, Alrafiah AR, et al. Current developments in gene therapy for amyotrophic lateral sclerosis. Expert Opin Biol Ther 2015; 15: 935-947.
- Lunn JS, Sakowski SA, Feldman EL. Concise review: stem cell therapies for amyotrophic lateral sclerosis: recent advances and prospects for the future. Stem Cells 2014; 32: 1099-1109.
- Richard JP, Maragakis NJ. Induced pluripotent stem cells from ALS patients for disease modeling. Brain Res 2014; 1607: 15-25.
Linked content
-
Podcast with Dr Thanuja Dharmadasa
Provenance: Not commissioned; externally peer reviewed.