




Helicobacter pylori infection is a major cause of morbidity and mortality worldwide. Infection invariably causes active chronic gastritis. In many people this may be clinically silent throughout life, but in a significant minority it results in gastroduodenal diseases, especially peptic ulcer disease, non-cardia gastric cancer and gastric mucosa-associated lymphoid tissue (MALT) lymphoma. H. pylori infection increases the risk of ulceration and bleeding in patients taking non-steroidal anti-inflammatory drugs (NSAIDs), including aspirin, and is responsible for symptoms in a subset of patients with functional dyspepsia. The seminal recognition of H. pylori by Marshall and Warren1 in 1982 was acknowledged by the award of the Nobel Prize in Physiology or Medicine in 2005.
Prevalence of Helicobacter pylori infection in Australia and New Zealand
It is estimated that more than 50% of the world’s population is infected with H. pylori.2 However, significant differences in the prevalence of infection exist within and between countries. In general, people in developing nations and residents of developed countries with low socio-economic status have a higher prevalence of infection.2-4
In most populations, including in Australia and New Zealand, the prevalence of H. pylori infection increases with age (Appendix).5-10 While acquisition of infection was initially believed to occur throughout life, it is now accepted that the increase in prevalence of H. pylori infection with increasing age reflects the passage through the population of distinct age cohorts whose acquisition rates of H. pylori in childhood were higher than those today. For example, in a study conducted in 2002, the prevalence of H. pylori infection in children aged 1–4 years was 4.0%, while the prevalence progressively increased in subsequent age cohorts, up to 23.3% in those aged 50–59 years.6 Given that acquisition occurs in childhood, these results and those of other studies in Australia indicate that the prevalence of H. pylori infection is decreasing in Australia (Appendix).
Based on studies conducted between 1988 and 2006, the overall prevalence of H. pylori infection in asymptomatic non-Indigenous Australians ranged from 15.4% to 30.6% (Appendix).6,11 In contrast, Indigenous Australian populations and migrant populations resident in Australia have significantly higher prevalence levels of H. pylori infection (Appendix).12,13 In a study conducted in Western Australia, not only was the prevalence of H. pylori infection significantly higher in Indigenous populations (76%) than in non-Indigenous populations (30%), but differences in the prevalence of infection between rural and urban Indigenous populations (91% v 60%) were observed.12 Similarly, residents of Australia born in high prevalence countries have been reported to have higher rates of infection than those born in Australia. For example, in one study of Australian residents, the overall prevalence of infection in adults born in western Europe, the United States and Canada (all 29.9%), southern Europe (51.3%), southern Africa and South America (both 46.2%) was higher than for those born in Australia or New Zealand (18.3%).13 Children born in Australia to parents who were born in countries with a high prevalence of infection also have higher prevalence rates.14 Further, extremely high prevalence rates of H. pylori infection have been reported in African refugee children in WA, ranging from 69% in those aged < 5 years to 91% in those aged > 10 years.15
A systematic review of studies conducted in New Zealand that investigated the prevalence of H. pylori infection in asymptomatic adults from four birth cohorts (1926–1940, 1941–1955, 1956–1970 and 1971–1985) of Maori, Pacific and European participants found the prevalence in Pacific Islanders to be significantly higher than that in Europeans.16
Origin and natural history of infection
Current evidence suggests that the close association between humans and H. pylori originated in Africa. It has been estimated that H. pylori has been associated with humans for the past 88 000–116 000 years, with acquisition of H. pylori infection possibly occurring by a host jump from an unknown source.17
Once established within the gastric mucosa of its host, H. pylori persists for life unless the infection is treated with antibiotics.2 In the early years of life, before infection is established, transient infection with H. pylori may be common; however, acquisition and loss of infection appear to differ in children from different ethnic backgrounds. For example, in a study involving white and African American children matched for socio-economic class, loss of infection over a 12-year period was significantly higher in the white children (50%) than the African American children (4%), the latter group remaining infected or becoming reinfected.2,18 True reinfection with H. pylori after successful eradication is uncommon in adults, the rate of reacquisition being reported as < 1% in many developed and some developing countries.19 In contrast, in other developing countries, it has been reported to be > 10%.19 However, this high rate is commonly due to recrudescence, in which antibiotic therapy suppresses, rather than eradicates, the H. pylori infection. In this scenario, after treatment is ceased, the number of bacteria increase over time, leading to recurrence of symptoms.
Transmission of infection
Transmission of H. pylori infection predominantly occurs from person to person within the family setting, with mothers playing a key role in transmitting H. pylori infection to their children.2,3 Evidence supporting this view comes from epidemiological studies showing that children with an infected mother have an increased risk of infection, and from studies comparing the genetic make-up of H. pylori strains present in the index child and his or her parents. For example, a Japanese study found that for children with H. pylori-positive mothers, the relative risk of acquiring infection was 5.3 times that of children whose mothers were H. pylori-negative.20 Further evidence from Japan showed that in 60% of children, the genetic make-up of H. pylori strains isolated from index paediatric patients matched the strains in their mothers, while 27.5% matched the genetic make-up of both parents.21
Although controversial, a further possible risk factor for person-to-person transmission is attendance at day care centres. A recent study reported the prevalence of H. pylori infection in Portuguese children attending day care centres for more than 3 years to be significantly increased (40.2%) compared with children who had never attended day care centres (13.2%).22 This finding is supported by a systematic review and meta-analysis of 16 studies, which showed the frequency of child care attendance to be a risk factor for H. pylori infection, particularly in settings with a high prevalence of infection.23
While investigations into the role of breastfeeding in the acquisition of H. pylori infection have produced conflicting results, a systematic review of epidemiological studies conducted between 1984 and 2007 reported breastfeeding to be protective against H. pylori infection, particularly in low and middle income countries.24
The most controversial area of H. pylori epidemiological research relates to its route of transmission. Given that H. pylori infection is located in the stomach and that H. pylori has a basic need for gastric-type mucosa for in vivo proliferation, ingestion appears to be the most likely means of acquisition. However, whether H. pylori reaches the oral cavity via the gastro–oral, oral–oral or faecal–oral route remains unclear.2,3
Currently, any role for water, pets or houseflies as possible transmission routes for H. pylori remains unproven.2,3,25
Risk factors associated with acquisition of Helicobacter pylori infection
Studies conducted in both developed and developing countries have identified low socio-economic status as being strongly associated with the acquisition of H. pylori infection, with socio-economic status during childhood being of particular importance.2-4 Socio-economic status encompasses a range of factors, including density of living, level of hygiene, sanitation and educational opportunities. In particular, low levels of education, high density of living, lack of sanitation and low hygiene levels are reported to increase the acquisition of infection within a population.2-4
This view is supported by the high prevalence rates reported in socio-economically disadvantaged Indigenous Australian communities12 and by the finding that Australians classified in the lowest quartile of socio-economic status (Q1) have a significantly higher prevalence of H. pylori infection (32.9%) than those in higher socio-economic quartiles (Q2–4: 18.4%, 19.9% and 20.8% respectively).13
Number of siblings and household crowding are also significant predictors of H. pylori infection.13 In New Zealand, household crowding among children born between 1971 and 1985 was shown to contribute to 44% of H. pylori infections in Pacific Islanders, 36% in Maori people and 14% in Europeans.16
Clinical impacts of Helicobacter pylori infection
Gastric cancer and MALT lymphoma
The risk of developing intestinal-type gastric adenocarcinoma depends on several interacting factors, including H. pylori virulence, H. pylori genetic ancestries, host genetics, dietary factors, essential micronutrients and the gastrointestinal microbiota.26 In susceptible infected hosts, active chronic gastritis results in gastric mucosal atrophy with intestinal metaplasia. In a small proportion of people, these pre-malignant mucosal changes lead to dysplasia and early (clinically silent), then advanced, gastric cancer. Gastric cancer mostly presents at an advanced, symptomatic stage and is associated with a poor prognosis. H. pylori infection has been estimated to confer an individual lifetime risk of gastric cancer of 1%–2%, irrespective of the population prevalence.27 Higher risk of gastric cancer relates to a greater risk of infection and genetic susceptibility. In Australia, this includes migrants from high prevalence areas, older people and those with a family history of gastric cancer. Uncomplicated longstanding dyspepsia is not an indication of the presence of gastric cancer. New onset of symptoms at an older age or alarm symptoms (including unexplained iron deficiency anaemia, overt bleeding, weight loss or dysphagia) at any age are indications for prompt endoscopy. However, if these symptoms are due to gastric cancer, they usually signify advanced disease with a poor prognosis.
Some well resourced countries with a high prevalence of gastric cancer conduct population screening for H. pylori infection and adverse gastric histology.28 In Australia, where other cancers are more common, population screening for H. pylori infection or precursor mucosal changes is not advocated, although case-by-case selection of individuals at higher risk (eg, those with a family history of gastric cancer) is appropriate. When gastric mucosal atrophy and intestinal metaplasia have been identified, endoscopic surveillance may be of benefit in individual cases, but an overall reduction in mortality is yet to be demonstrated. When dysplasia is found, focal areas of high-grade dysplasia may be removed endoscopically, while more extensive changes require surgery.
Gastric MALT lymphoma is a rare manifestation of H. pylori infection. Eradication of H. pylori when the lymphoma is at a low grade stage usually results in regression and cure.29
Peptic ulcer disease
H. pylori infection has been shown to cause most duodenal ulcers and about two-thirds of gastric ulcers. However, with decreasing prevalence of infection in Australia and increasing cure of ulcer patients, the proportion of all peptic ulcers due to H. pylori is falling. Without definitive treatment, peptic ulcer disease is a chronic relapsing and remitting disease that causes major morbidity and mortality from pain, bleeding and perforation. Eradication of H. pylori infection heals most active peptic ulcers and prevents further relapse, thus effectively curing the disease. For complicated ulcer disease (such as bleeding ulcers) or large gastric ulcers that may be slow to heal even after eradication of H. pylori infection, a short course of proton pump inhibitor (PPI) therapy is usually also given. Additional treatment with a PPI is not usually required for uncomplicated duodenal ulcer disease, as long as successful eradication is confirmed. Asymptomatic patients with a past history of peptic ulcer disease should be tested for H. pylori infection and treated to prevent further relapses.
NSAIDs, including aspirin, cause most other ulcer disease and an increasing proportion of all ulcers. H. pylori and NSAIDs act synergistically to increase the risk of ulcers and bleeding.30 As eradication of H. pylori infection markedly reduces this risk,31 guidelines advocate testing for and treating H. pylori infection before commencing chronic NSAID therapy.32 When a patient with an ulcer who is taking NSAIDs is found to also have H. pylori infection, both risk factors must be addressed. Following ulcer bleeding and when NSAIDs are still required, eradication therapy is appropriate, but PPIs are also needed to reduce the risk of recurrent bleeding. Low dose aspirin use increases the risk of peptic ulceration and bleeding, and H. pylori infection adds to this risk. In patients known to have previously had ulcer disease or bleeding, H. pylori should be tested for and treated before aspirin is used.33
H. pylori-associated dyspepsia
H. pylori gastritis is commonly associated with upper gut symptoms. However, only up to a third of infected patients with functional dyspepsia will have sustained relief of symptoms after eradication therapy, because functional dyspepsia is a heterogeneous condition. H. pylori may be causal in some patients and incidentally present in others. However, the proportion of infected patients whose symptoms abate after eradication therapy is greater than for those given empirical acid suppression therapy.34 Furthermore, patients may benefit from a reduced lifetime risk of ulcer disease and cancer, especially if they are treated before adverse histological changes have developed in the gastric mucosa.30 For both reasons, it is recommended to offer these patients eradication therapy.28,32,35
A recent revised classification of gastritis has recognised H. pylori-associated dyspepsia as a distinct entity, and this will be incorporated into the 11th revision of the International Classification of Diseases.28 The classification also highlights the significance of H. pylori gastritis as the precursor lesion that leads to peptic ulcer disease and gastric cancer, irrespective of whether symptoms are present.
H. pylori infection has been associated with a variety of other conditions. In most cases, the association has not been shown to be causal, and common conditions will inevitably coexist in some patients. There are modest data linking H. pylori to immune thrombocytopenic purpura, and eradication therapy is often tried, with variable results.35
Diagnosis
The choice of diagnostic test depends on the clinical context. For uncomplicated upper gut symptoms in younger patients, a non-invasive “test and treat” strategy using the stable carbon isotope 13 (13C) or radioactive carbon 14 (14C) urea breath test is appropriate, with recourse to endoscopy if this strategy fails to relieve symptoms.32 Validated breath tests are highly sensitive and specific. Tests with indeterminate results should be repeated after several weeks. Faecal antigen tests have also been used but are less convenient and somewhat less accurate. Serology is the least sensitive and specific test and, while useful in epidemiological studies, is not considered sufficiently accurate for individual clinical decision making.36 Moreover, as antibodies may persist for years, serological testing is not appropriate to determine the outcome of therapy. When endoscopy is indicated, H. pylori infection is readily diagnosed by biopsy. Use of two diagnostic modalities, usually histology and a rapid urease test, with biopsy samples taken from two topographical locations in the stomach (antrum and corpus) maximises accuracy. Biopsy for culture is not used as a primary diagnostic test.
As failure of eradication therapy is not uncommon,37 the outcome of therapy should be assessed. This is usually done with a urea breath test not less than 4 weeks after cessation of therapy. When endoscopy is required, such as to ensure the healing of gastric ulcers and to exclude neoplasia, the outcome of therapy may be assessed using repeat biopsy samples taken at that time. The finding of active chronic gastritis without any organisms identified should prompt suspicion for undetected infection and further testing.
False negative results from urea breath tests, faecal antigen tests and biopsies may occur when there has been recent antibiotic use (within 4 weeks) or recent use of PPIs (within 1–2 weeks). Hence, when possible, PPIs should be withheld before testing to maximise diagnostic accuracy.38
Treatment and drug resistance
The decision to treat or not to treat H. pylori infection must be an active one, taking an individual patient’s circumstances and risks into consideration. The decision to test for H. pylori infection is therefore made with therapeutic intent. Recommended indications for treatment are summarised in Box 1.
The major determinant of successful eradication is the presence or absence of pre-treatment antimicrobial resistance. Lack of adherence with treatment and smoking are also factors.40 The choice of therapy is based on local antibiotic usage, documented antibiotic resistance and outcome data (Box 2). Thus, recommended therapies will vary regionally. The most widely used and recommended treatment (including in Australia) is triple therapy comprising a PPI, amoxycillin and clarithromycin, usually for 1 week.41 In countries where clarithromycin has been widely used as monotherapy for other infections, resistance to it is often high (> 20%), which greatly reduces the effectiveness of this therapy — often to below the acceptable benchmark eradication rate of > 80%. This has led to calls to abandon this therapy as first-line treatment and instead use a variety of other combinations and dosing. These so-called concomitant, sequential or hybrid regimens combine these three drugs with a nitroimidazole or other agents and increase the duration of therapy to 10–14 days. However, in Australia, clarithromycin resistance has been repeatedly documented to be low (6%–8%),37,42 so this advice may not be relevant to the Australian situation and has not been tested locally. Fortunately, resistance to amoxycillin is virtually non-existent and does not develop after treatment failure. Conversely, metronidazole resistance in Australia is known to be very high (45%–50%), almost certainly due to widespread, long term use of nitroimidazoles as monotherapy.43 Therefore, clarithromycin remains an effective component of triple therapy in Australia, while adding metronidazole to this combination will add adverse effects, adherence difficulties and cost, while adding only modestly to predicted outcomes. Such a regimen has not been formally tested in Australia, and studies from abroad vary in quality and outcomes. The option of extending the duration of standard triple therapy has similarly not been tested in Australia, but such studies are needed as there is anecdotal evidence that current local results do not match those from randomised controlled trials published years ago.
For patients who are allergic to penicillin (Box 3), metronidazole may be substituted for amoxycillin, although pre-treatment metronidazole resistance reduces the efficacy of this regimen. A penicillin-free, bismuth-containing quadruple therapy may be used instead (see below). Alternatively, formal penicillin allergy testing may be done for the many patients with a remote or unlikely history of allergy. Up to 80% of such patients have been found not to be allergic to penicillin and may be treated with standard therapy.44
The main role of culturing endoscopic biopsy samples is to monitor changes in antimicrobial resistance over time. When therapy fails, individual antibiotic sensitivity testing from cultured biopsy samples is seldom helpful, as it has little role in clinical decision making. Secondary clarithromycin resistance frequently follows failure of first-line therapy and is therefore assumed in this scenario. Repeat treatment with a clarithromycin-containing combination has a very low rate of success (about 10%). Moreover, in vitro sensitivity of other antibiotics does not infer in vivo efficacy, and ad hoc regimens designed in this way should also be avoided.
Proven second-line or salvage eradication therapies are well documented, but some of the components of these combinations are not registered for use and are not readily available in Australia. They may be obtained from abroad after approval from the Therapeutic Goods Administration through the Special Access Scheme, but second-line therapy should remain the domain of the interested and informed practitioner. In Australia, three evidence-based options for second-line therapy have been evaluated (Box 2). Levofloxacin-based triple therapy (PPI, amoxycillin and levofloxacin) has been shown to result in high eradication rates irrespective of the number of prior treatment failures.45 Quadruple therapy of bismuth subcitrate, a PPI, tetracycline and metronidazole, while clumsy in terms of dosage, is also effective, as metronidazole resistance is overcome by this four-drug regimen.37 Rifabutin-based triple therapy may also be used but is less effective, and the occasional occurrence of neutropenia tends to limit its use.46 These combinations may be used sequentially for repeated treatment failures, if necessary. In experienced centres, final eradication rates with judiciously chosen therapy should approach 99%.47
Box 1 – Indications for Helicobacter pylori infection eradication therapy*
Indication |
Benefits of treatment |
||||||||||||||
Past or present peptic ulcer disease |
Heals ulcers and reduces relapse |
||||||||||||||
Functional dyspepsia |
May reduce symptoms and long term risk of ulcer disease and gastric cancer |
||||||||||||||
Use of NSAIDs, including aspirin, in some patients |
Reduces risk of ulcers and bleeding |
||||||||||||||
Gastric mucosal atrophy and intestinal metaplasia |
Reduces risk of gastric cancer |
||||||||||||||
Patients requiring long term acid suppression with proton pump inhibitors |
Reduces progression of intestinal metaplasia |
||||||||||||||
Family history of gastric cancer |
Reduces long term risk of gastric cancer |
||||||||||||||
Prior early gastric cancer |
Reduces risk of further gastric cancer |
||||||||||||||
Low grade gastric MALT lymphoma |
Induces regression of lymphoma |
||||||||||||||
Patient choice, after risks and benefits are discussed |
Fulfils patient’s desire to reduce risk of infection |
||||||||||||||
Strategy for gastric cancer prevention in communities with high incidence of gastric cancer |
In Australia, may help subsets of people at high risk |
||||||||||||||
NSAIDs = non-steroidal anti-inflammatory drugs. MALT = mucosa-associated lymphoid tissue. * Adapted from Fock KM, et al. J Gastroenterol Hepatol 2009; 24: 1587-1600.39 | |||||||||||||||
Box 2 – Treating Helicobacter pylori infection when primary clarithromycin resistance rate is low and metronidazole resistance rate is high*
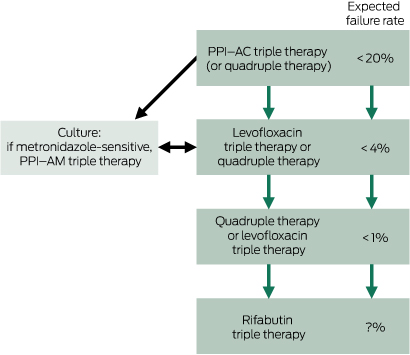
PPI = proton pump inhibitor. PPI–AC = PPI, amoxycillin, clarithromycin. PPI–AM = PPI, amoxycillin, metronidazole. Levofloxacin triple therapy = PPI, amoxycillin, levofloxacin. Quadruple therapy = PPI, bismuth subsalicylate, tetracycline, metronidazole. Rifabutin triple therapy = PPI, amoxycillin, rifabutin. * See text for details of regimens. For doses, duration and availability, see Therapeutic guidelines: gastrointestinal. Version 5.41
Box 3 – Treating Helicobacter pylori infection in patients with a history of possible penicillin allergy*
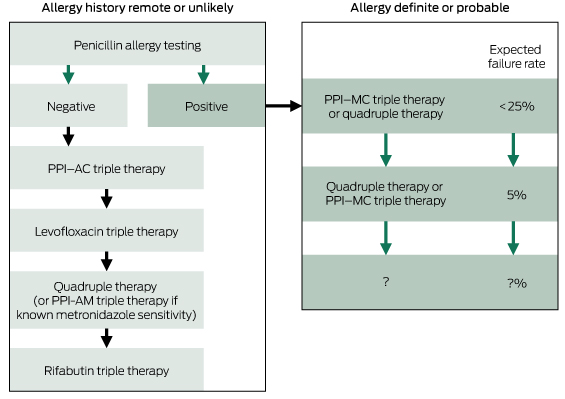
PPI = proton pump inhibitor. PPI–AC = PPI, amoxycillin, clarithromycin. PPI–AM = PPI, amoxycillin, metronidazole. Levofloxacin triple therapy = PPI, amoxycillin, levofloxacin. Quadruple therapy = PPI, bismuth subsalicylate, tetracycline, metronidazole. Rifabutin triple therapy = PPI, amoxycillin, rifabutin. PPI–MC = PPI, metronidazole, clarithromycin. * See text for details of regimens. For doses, duration and availability, see Therapeutic guidelines: gastrointestinal. Version 5.41 * Katelaris P, Katelaris A. J Gastroenterol Hepatol 2015; 30 Suppl 3: 178.44
Provenance: Commissioned; externally peer reviewed.
- Hazel Mitchell1
- Peter Katelaris2
- 1 School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW
- 2 Department of Gastroenterology, University of Sydney, Sydney, NSW
* Equal first authors
No relevant disclosures.
- 1. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984; 1: 1311-1315.
- 2. Mitchell HM. Epidemiology of infection. In: Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: physiology and genetics. Washington, DC: ASM Press, 2001.
- 3. Sierra MS, Hastings EV, Fagan-Garcia K, et al. Epidemiology, transmission and public health implications of Helicobacter pylori infection in Western countries. In: Buzas GM, editor. Helicobacter pylori — a worldwide perspective. Bentham Science Publishers, 2014: 25-79.
- 4. Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter 2014; 19 Suppl 1: 1-5.
- 5. Mitchell HM, Lee A, Berkowicz J, Borody T. The use of serology to diagnose active Campylobacter pylori infection. Med J Aust 1988; 149: 604-609.
- 6. Moujaber T, MacIntyre CR, Backhouse J, et al. The seroepidemiology of Helicobacter pylori infection in Australia. Int J Infect Dis 2008; 12: 500-504.
- 7. Fawcett JP, Shaw JP, Cockburn M, et al. Seroprevalence of Helicobacter pylori in a birth cohort of 21-year-old New Zealanders. Eur J Gastroenterol Hepatol 1996; 8: 365-369.
- 8. Fraser AG, Scragg R, Metcalf P, et al. Prevalence of Helicobacter pylori infection in different ethnic groups in New Zealand children and adults. Aust N Z J Med 1996; 26: 646-651.
- 9. Fraser AG, Scragg R, Schaaf D, et al. Helicobacter pylori infection and iron deficiency in teenage females in New Zealand. N Z Med J 2010; 123: 38-45.
- 10. Hsiang JJ, Selvaratnam S, Taylor S, et al. Increasing primary antibiotic resistance and ethnic differences in eradication rates of Helicobacter pylori infection in New Zealand — a new look at an old enemy. N Z Med J 2013; 126: 64-76.
- 11. Peach HG, Pearce DC, Farish SJ. Helicobacter pylori infection in an Australian regional city: prevalence and risk factors. Med J Aust 1997; 167: 310-313. <MJA full text>
- 12. Windsor HM, Morrow SD, Marshall BM, et al. Prevalence of Helicobacter pylori in Indigenous Western Australians: comparison between urban and remote rural populations. Med J Aust 2005; 182: 210-213. <MJA full text>
- 13. Pandeya N, Whiteman DC; Australian Cancer Study. Prevalence and determinants of Helicobacter pylori sero-positivity in the Australian adult community. J Gastroenterol Hepatol 2011; 26: 1283-1289.
- 14. Hardikar W, Grimwood K. Prevalence of Helicobacter pylori infection in asymptomatic children. J Paediatr Child Health 1995; 31: 537-541.
- 15. Cherian S, Forbes D, Sanfilippo F, et al. The epidemiology of Helicobacter pylori infection in African refugee children resettled in Australia. Med J Aust 2008; 189: 438-441. <MJA full text>
- 16. McDonald AM, Sarfati D, Baker M, Blakely T. Trends in Helicobacter pylori infection among Maori, Pacific and European birth cohorts in New Zealand. Helicobacter 2015; 20: 139-145.
- 17. Moodley Y, Linz B, Bond RP, et al. Age of the association between Helicobacter pylori and man. PLoS Pathog 2012; 8: e1002693. doi: 10.1371/journal.ppat.1002693.
- 18. Malaty HM, Graham DY, Wattigney WA, et al. Natural history of Helicobacter pylori infection in childhood: 12-year follow-up cohort study in a biracial community. Clin Infect Dis 1999; 28: 279-282.
- 19. Zhang YY, Xia HH, Zhuang ZH, Zhong J. Review article: ‘true’ re-infection of Helicobacter pylori after successful eradication — worldwide annual rates, risk factors and clinical implications. Aliment Pharmacol Ther 2009; 29: 145-160.
- 20. Malaty HM, Kumagai T, Tanaka E, et al. Evidence from a nine-year birth cohort study in Japan of transmission pathways of Helicobacter pylori infection. J Clin Microbiol 2000; 38: 1971-1973.
- 21. Yokota S, Konno M, Fujiwara S, et al. Intrafamilial, preferentially mother-to-child and intraspousal, Helicobacter pylori infection in Japan determined by mutilocus sequence typing and random amplified polymorphic DNA fingerprinting. Helicobacter 2015; 20: 334-342.
- 22. Lunet N, Peleteiro B, Bastos J, et al. Child day-care attendance and Helicobacter pylori infection in the Portuguese birth cohort Geracao XXI. Eur J Cancer Prev 2014; 23: 193-198.
- 23. Bastos J, Carreira H, La Vecchia C, Lunet N. Childcare attendance and Helicobacter pylori infection: systematic review and meta-analysis. Eur J Cancer Prev 2013; 22: 311-319.
- 24. Chak E, Rutherford GW, Steinmaus C. The role of breast-feeding in the prevention of Helicobacter pylori infection: a systematic review. Clin Infect Dis 2009; 48: 430-437.
- 25. Allen SJ, Thomas JE, Alexander ND, et al. Flies and Helicobacter pylori infection. Arch Dis Child 2004; 89: 1037-1038.
- 26. Amieva M, Peek RM Jr. Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology 2016; 150: 64-78.
- 27. Chang AH, Parsonnet J. Role of bacteria in oncogenesis. Clin Microbiol Rev 2010; 23: 837-857.
- 28. Sugano K, Tack J, Kuipers EJ, et al; faculty members of Kyoto Global Consensus Conference. Kyoto global consensus report on Helicobacter pylori gastritis. Gut 2015; 64: 1353-1367.
- 29. Fischbach W. Gastric mucosal-associated lymphoid tissue lymphoma. Gastroenterol Clin North Am 2013; 42: 371-380.
- 30. Huang JQ, Sridhar S, Hunt RH. Role of Helicobacter pylori infection and non-steroidal anti-inflammatory drugs in peptic-ulcer disease: a meta-analysis. Lancet 2002; 359: 14-22.
- 31. Chan FK, To KF, Wu JC, et al. Eradication of Helicobacter pylori and risk of peptic ulcers in patients starting long-term treatment with non-steroidal anti-inflammatory drugs: a randomised trial. Lancet 2002; 359: 9-13.
- 32. Malfertheiner P, Megraud F, O’Morain CA, et al; European Helicobacter Study Group. Management of Helicobacter pylori infection — the Maastricht IV/ Florence Consensus Report. Gut 2012; 61: 646-664.
- 33. Chan FK, Chung SC, Suen BY, et al. Preventing recurrent upper gastrointestinal bleeding in patients with Helicobacter pylori infection who are taking low-dose aspirin or naproxen. N Engl J Med 2001; 344: 967-973.
- 34. Chiba N, Van Zanten SJ, Sinclair P, et al. Treating Helicobacter pylori infection in primary care patients with uninvestigated dyspepsia: the Canadian adult dyspepsia empiric treatment-Helicobacter pylori positive (CADET-Hp) randomised controlled trial. BMJ 2002; 324: 1012-1016.
- 35. Frydman GH, Davis N, Beck PL, Fox JG. Helicobacter pylori eradication in patients with immune thrombocytopenic purpura: a review and the role of biogeography. Helicobacter 2015; 20: 239-251.
- 36. Loy CT, Irwig LM, Katelaris PH, Talley NJ. Do commercial serological kits for Helicobacter pylori infection differ in accuracy? A meta-analysis. Am J Gastroenterol 1996; 91: 1138-1144.
- 37. Katelaris PH, Forbes G, Talley NJ, Crotty B. A randomized comparison of quadruple and triple therapies for Helicobacter pylori eradication: the QUADRATE Study. Gastroenterology 2002; 123: 1763-1769.
- 38. Connor SJ, Seow F, Ngu MC, Katelaris PH. The effect of dosing with omeprazole on the accuracy of the 13C-urea breath test in Helicobacter pylori-infected subjects. Aliment Pharmacol Ther 1999; 13: 1287-1293.
- 39. Fock KM, Katelaris P, Sugano K, et al. Second Asia-Pacific Consensus Guidelines for Helicobacter pylori infection. J Gastroenterol Hepatol 2009; 24: 1587-1600.
- 40. Suzuki T, Matsuo K, Ito H, et al. Smoking increases the treatment failure for Helicobacter pylori eradication. Am J Med 2006; 119: 217-224.
- 41. Gastrointestinal Expert Group. Gastric disorders. In: Therapeutic guidelines: gastrointestinal. Version 5. Melbourne: Therapeutic Guidelines Ltd, 2011.
- 42. Katelaris PH, Adamthwaite D, Midolo P, et al. Randomised trial of omeprazole and metronidazole with amoxycillin or clarithromycin for Helicobacter pylori eradication, in a region of high primary metronidazole resistance: the HERO study. Aliment Pharmacol Ther 2000; 14: 751-758.
- 43. Katelaris PH, Nguyen TV, Robertson GJ, et al. Prevalence and demographic determinants of metronidazole resistance by Helicobacter pylori in a large cosmopolitan cohort of Australian dyspeptic patients. Aust N Z J Med 1998; 28: 633-638.
- 44. Katelaris P, Katelaris A. The role of immunological testing for optimising treatment of patients with refractory Helicobacter pylori infection and a history of penicillin allergy [abstract]. J Gastroenterol Hepatol 2015; 30 Suppl 3: 178.
- 45. Katelaris PH, Maseeh A, Tattersall SJN, Katelaris AL. A prospective evaluation of levofloxacin based triple therapy for refractory H. pylori infection in a large Australian cohort [abstract PP43]. J Gastroenterol Hepatol 2012; 27 Suppl 1: 16.
- 46. Van der Poorten D, Katelaris PH. The effectiveness of rifabutin triple therapy for patients with difficult-to-eradicate Helicobacter pylori in clinical practice. Aliment Pharmacol Ther 2007; 26: 1537-1542.
- 47. Gisbert JP. “Rescue” regimens after Helicobacter pylori treatment failure. World J Gastroenterol 2008; 14: 5385-5402.
Summary