





Lessons from practice
Volume 202 - Issue 2
Vasculitis or fibromuscular dysplasia?
Authors: Michelle C Papandony, Sharmayne RE Brady and Tai-Juan Aw
Med J Aust 2015; 202 (2): 100-101. || doi: 10.5694/mja14.00224
Published online: 2 February 2015
Published online: 2 February 2015
A 45-year-old previously well woman with a probable diagnosis of Takayasu arteritis
Clinical record
A 45-year-old previously well woman of Filipina descent was admitted to hospital following an episode of vertigo that spontaneously resolved after 2 minutes. The patient denied experiencing any associated symptoms. Her past history included hypertension, diagnosed when she was 27 years old. She did not smoke or drink alcohol. Her brother suffered a stroke at age 46 years.
On physical examination, the patient's blood pressure was 160/100 mmHg in the right arm and 140/95 mmHg in the left arm. Her blood glucose level was 6.8 mmol/L (reference interval [RI], 4–11 mmol/L). There were loud bruits in both carotid arteries and the left renal artery. The right radial pulse was stronger than the left. Both lower limb pulses were palpable. Results of the remainder of the physical examination were normal.
Results of a full blood count, tests for urea and electrolyte levels, liver function tests, thyroid function tests, and tests for troponin and C-reactive protein levels were normal. Erythrocyte sedimentation rate was slightly elevated at 22 mm/h (RI, < 20 mm/h). Results of vasculitis screening — tests for antinuclear antibodies, extractable nuclear antigens, antibodies to double-stranded DNA and antineutrophil cytoplasmic antibodies — were normal. Results of thrombophilia screening — tests for anticardiolipin IgG, antithrombin III, protein C function, free protein S, prothrombin gene (20210) mutation, factor V Leiden and anti-β2 glycoprotein 1 antibodies — were also normal. Total cholesterol level was 2.6 mmol/L (RI, < 5.5 mmol/L), low-density lipoprotein level was 1.5 mmol/L (RI, < 3.5 mmol/L) and high-density lipoprotein was 0.9 mmol/L (RI, > 1.0 mmol/L).
A computed tomography (CT) scan of the brain was unremarkable. Magnetic resonance imaging of the brain showed supratentorial T2-weighted and fluid-attenuated inversion recovery (FLAIR) white matter hyperintensity, suggesting moderate chronic small-vessel ischaemic disease. Carotid and vertebral duplex ultrasound confirmed high-grade stenosis in both internal and external carotid arteries. CT angiography showed extensive atherosclerosis for the patient's age and tight stenosis in the right internal carotid artery and distal aspect of the left common carotid artery. CT angiography of the thoracic aorta revealed a descending aortic aneurysm and thrombosis of the left subclavian artery. A CT renal angiogram revealed a stenosis at the origin of the left renal artery (Figure, arrow). A full body positron emission tomography scan could not be interpreted because of physiological uptake of contrast medium by brown fat, which limited the ability to evaluate for large-vessel vasculitis.
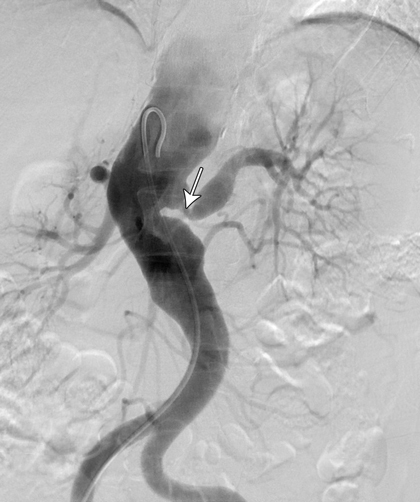
The combination of both stenotic and aneurysmal changes in multiple vascular beds makes Takayasu arteritis (TA) a compelling diagnosis in this patient. Given the absence of disease activity, late-stage TA is a probable diagnosis. Patients with fibromuscular dysplasia (FMD) can also present in this manner, but FMD is unlikely to involve several vascular territories (Box).
TA is an inflammatory vasculopathy of the aorta and its main branches.1 It was once thought to be a disorder that affected only Asian women, but it has since been identified in men and women from many ethnic groups,2 usually in the second or third decade of life.3
The pathogenesis of TA is poorly understood. Vessel inflammation results in wall thickening, fibrosis, stenosis and thrombus formation.2 Panarteritis with adventitial and intimal scarring extends distally from the aorta into its branches, causing narrowing and occlusions. Inflammation can destroy the arterial media and lead to aneurysm formation.2 Morbidity due to vascular occlusion and aneurysm formation is high.
TA has a triphasic pattern of disease progression. The clinical features vary depending on the degree of arterial dilation, narrowing and occlusion. Phase I is the inflammatory period, which is characterised by non-specific symptoms such as fever and weight loss. Phase II involves vessel tenderness due to inflammation. Phase III is the burned-out (or late) stage, in which bruits and ischaemia predominate in multiple organs.4 Phase III does not occur in all patients. In patients who are deemed to be in remission, relapses can occur.
FMD is an uncommon disease that mimics vasculitis. It is a non-inflammatory, non-atherosclerotic angiopathy that occurs most commonly in women of child-bearing age.5 It has been observed in nearly every arterial bed.
The clinical manifestations of FMD depend on the arterial distribution affected. FMD may cause stenosis, aneurysm, dissection and occlusion of arteries.6 Common manifestations include dizziness, hypertension and stroke.7 Patients with FMD commonly have involvement of the renal and carotid arteries.7,8 FMD has three subtypes, according to the dominant arterial wall layer involved.
There are instances when it is difficult to differentiate FMD from vasculitis. As FMD is a non-inflammatory process, it is not associated with anaemia, thrombocytopenia or elevated levels of acute phase reactants.9 If histological proof of FMD or inflammation is not available, distinguishing between these conditions may be difficult because the angiographic appearances are similar.9
Diagnostic confirmation for our patient could only be achieved with arterial biopsy. This was not an option owing to procedural risk. Given the number of vascular beds involved, TA was the favoured diagnosis. The absence of constitutional symptoms and low levels of inflammatory markers suggested that late-phase TA was likely, but active TA could not be excluded. Distinguishing between these entities is clinically important as the therapy differs.
The management of patients with TA can be challenging. The lack of a single gold standard measure of disease activity often results in the diagnosis being established from a combination of clinical history, pathology tests and imaging. There may be poor correlation between the results of such investigations, leading to uncertainty regarding diagnosis and appropriate therapy.
The goal of treatment in TA is to minimise long-term adverse effects while controlling disease activity and preserving vascular competence. In active TA, this is achieved with corticosteroids and cytotoxic medications. Use of these medications in late-phase TA has been contentious. The lack of a measurable inflammatory response may cause uncertainty regarding whether to aggressively immunosuppress the patient. Despite this, patients who are deemed to have late-phase TA may still have histological evidence of ongoing disease. Moreover, although the 10-year mortality is low, morbidity due to arterial occlusion is high in TA.10 Since inflammation is a risk factor for atherosclerosis, traditional cardiovascular risk factors should be screened for routinely and managed with antiplatelet agents, antihypertensive medication and statin therapy, as deemed necessary. However, using angiotensin-converting enzyme inhibitors requires close monitoring in view of the prevalence of renal artery stenosis. Stenting and balloon angioplasty can be used to maintain vascular patency.
TA is a challenging disease to diagnose and manage. Identification of superior diagnostic tools, including imaging techniques, may facilitate our understanding of disease activity and guide therapeutic interventions. Given a shortage of evidence regarding therapy in late-phase TA, patients should be assessed on a case-by-case basis and those who have a good prognosis should not be put at risk by treatment that is more harmful than the disease itself. Ultimately, a multidisciplinary approach with long-term follow-up is needed.
Lessons from practice
- Fibromuscular dysplasia can mimic Takayasu arteritis.
- Various factors, including age, vascular territory involvement and inflammatory markers, can help distinguish Takayasu arteritis from other diseases.
- Patients who have a good prognosis should not be put at risk by treatment that is more harmful than the disease itself.
- Secondary prevention and management of cardiovascular risk factors are vital in patients with Takayasu arteritis.
Differential diagnoses that were considered for a 45-year-old woman who had stenotic and aneurysmal changes in multiple vascular beds
Consistent with diagnosis | Less consistent with diagnosis | ||||||||||||||
Burned-out Takayasu arteritis | |||||||||||||||
|
| ||||||||||||||
Active Takayasu arteritis | |||||||||||||||
|
| ||||||||||||||
Fibromuscular dysplasia | |||||||||||||||
|
| ||||||||||||||
Atherosclerosis | |||||||||||||||
|
| ||||||||||||||
Giant cell arteritis | |||||||||||||||
|
| ||||||||||||||
Competing interests
No relevant disclosures.
References
- Liang PA, Hoffman GS. Advances in the medical and surgical treatment of Takayasu arteritis. Curr Opin Rheumatol 2005; 17: 16-24. 1
- Saadoun D, Lambert M, Mirault T, et al. Retrospective analysis of surgery versus endovascular intervention in Takayasu arteritis: a multicenter experience. Circulation 2012; 125: 813-819. 2
- Johnston SL, Lock RJ, Gompels MM. Takayasu arteritis: a review. J Clin Pathol 2002; 55: 481-486. 3
- Lupi-Herrera E, Sánchez-Torres G, Marcushamer J, et al. Takayasu's arteritis: clinical study of 107 cases. Am Heart J 1977; 93: 94-103. 4
- Slovut DP, Olin JW. Fibromuscular dysplasia. N Engl J Med 2004; 350: 1862-1871. 5
- Olin JW, Pierce M. Contemporary management of fibromuscular dysplasia. Curr Opin Cardiol 2008; 23: 527-536. 6
- Olin JW, Sealove BA. Diagnosis, management, and future development of fibromuscular dysplasia. J Vasc Surg 2011; 53: 826-836. 7
- Weinberg I, Jaff MR. Nonatherosclerotic arterial disorders of the lower extremities. Circulation 2012; 126: 213-222. 8
- Begelman SM, Olin JW. Fibromuscular dysplasia. Curr Opin Rheumatol 2000; 12: 41-47. 9
- Youngstein T, Mehta P, Mason J. Takayasu arteritis: a ten year longitudinal study in the UK [abstract]. Rheumatology 2011; 50 Suppl 3: iii146. lefthere