





Volume 201 - Issue 1
Skin rash, a kidney mass and a family mystery dating back to World War II
Authors: Christopher W Toon, Csilla Hasovits, Julie Paik, Michael Field, Angela Chou, Thomas J Hugh, Nicholas Pavlakis and Anthony J Gill
Med J Aust 2014; 201 (1): 58-60. || doi: 10.5694/mja13.00230
Published online: 7 July 2014
Published online: 7 July 2014
Consider a hereditary basis for renal cancer in young patients
This case of hereditary leiomyomatosis and renal cell cancer in a young man illustrates the importance of considering a hereditary basis for renal cancer in a young patient, and highlights how targeted therapy underpins modern personalised medicine in renal oncology.
Clinical record
A 25-year-old white man presented with a year-long history of malaise, a palpable right loin mass and a painless nodular rash over his back of indeterminate duration. Computed tomography (CT) showed a right renal mass with para-aortic and retroperitoneal lymphadenopathy. The patient underwent radical nephrectomy and para-aortic lymphadenectomy.
Examination of the kidney showed a 14 cm tumour centred in the renal medulla (Box, A). The tumour cells were organised in papillary fronds and featured abundant eosinophilic cytoplasm, large nuclei and prominent inclusion-like eosinophilic nucleoli (Box, B). There was widespread vascular space invasion and involvement of the renal sinus fat and hilar vein. All four para-aortic lymph nodes contained metastatic disease.
The patient's longstanding skin rash (Box, C) was reviewed and confirmed on biopsy to represent multiple cutaneous leiomyomata.1 The patient's mother was found to have had multiple cutaneous and uterine leiomyomata. The death certificate of his maternal grandfather stated that he died of metastatic renal carcinoma at the age of 44. He had returned from active service in World War II with a mysterious and longstanding rash, the pattern of which was consistent with cutaneous leiomyomatosis. On this basis a provisional diagnosis of hereditary leiomyomatosis and renal cell cancer (HLRCC) was made.1
After the patient underwent genetic counselling and provided informed consent, blood was drawn for molecular analysis of the fumarate hydratase (FH) gene locus by Sanger sequencing and multiplex ligation-dependent probe amplification assay (MLPA). MLPA showed complete deletion of the FH gene in one allele, confirmed by comparative genomic hybridisation on an Agilent custom oligonucleotide array (Agilent Technologies Inc) to be a 0.5 Mb deletion including FH and OPN3. No further cascade genetic testing has been undertaken in this family to date.
The patient's postoperative recovery was uneventful. Fluorodeoxyglucose positron emission tomography 6 months after surgery showed multiple hepatic and left flank chest wall subcutaneous metastatic deposits. There was no evidence of recurrence in the nephrectomy bed. First-line therapy with sunitinib (50 mg daily, orally) was commenced and well tolerated.
Eighteen months after the patient's surgery, CT imaging showed a reduction in the volume of the hepatic tumour and complete regression of the previously identified extra-hepatic disease. Subsequent planned subtotal hepatectomy was aborted due to the size and location of hepatic disease apparent only at exploratory laparotomy. Therapy was changed to everolimus (10 mg daily, orally). This resulted in symptomatic anaemia requiring transfusion. A restaging CT scan performed 8 months after commencement of therapy with everolimus showed progression in the size of the hepatic metastatic deposits.
Treatment was then changed to combination therapy with bevacizumab (15 mg/kg intravenous infusion every 3 weeks) and erlotinib (150 mg daily, orally) based on the treatment arm of a current National Cancer Institute phase II clinical trial (NCI-10-C-0114). The disease was stable for 16 months after commencement of the combination regimen, with the patient able to return to full-time employment. However he subsequently developed progressive disease and, despite a brief trial of sorafenib, died with widespread metastases 64 months after his initial presentation.
Discussion
At least 4% of renal cancers are hereditary, and there are known associations with germline mutations of VHL, c-MET, SDHx and FLCN loci and constitutional translocations of chromosome 3.2 Therefore, the possibility of hereditary disease should always be considered in people who present with renal carcinoma at a young age.
HLRCC is an autosomal dominant syndrome characterised by an inherited predisposition to cutaneous and uterine leiomyomata and renal cancer. Although multigenerational families with cutaneous and uterine leiomyomata were first described in the 1950s,3 the syndromic association with renal cancer was not appreciated until 2001 when two Finnish kindreds with a predisposition for cutaneous and uterine leiomyomata and early onset renal cancers were reported.4 Renal tumours occurred in four members of the index family in their 30s; all tumours were unilateral solitary lesions that had metastasised at the time of diagnosis, and displayed papillary architecture on histological examination. Using genome-wide linkage analysis, it was possible to map the genetic abnormality to the long arm of chromosome 1 (1q24). Subsequent studies have shown that this locus encodes FH, an enzyme involved in oxidative respiration through the Krebs cycle.5,6 The gene appears to function as a tumour suppressor in patients with HLRCC, in keeping with Knudson's two-hit hypothesis.7
The distinctive histological features of HLRCC renal tumours first described in 20014 have recently been corroborated in a review of 40 tumours resected from members of HLRCC-affected families. It was primarily that we recognised this distinct morphology (a papillary architecture with strikingly prominent nucleoli)1 that subsequently allowed us to make a definitive diagnosis by molecular confirmation in this case.
Given the rarity of HLRCC, no consensus diagnostic criteria currently exist for the condition. The presence of cutaneous leiomyomata correlates strongly with the presence of germline mutations in the FH gene.8 Cutaneous leiomyomata tend to occur more commonly in women, are typically sensitive to cold or abrasion, and develop in the second and fourth decades of life as intradermal lesions with a disseminated or segmental distribution.6 Although uterine leiomyoma is a common incidental finding in adulthood, the presence of multiple large symptomatic uterine leiomyomata further increases the likelihood of HLRCC. The histology of these cutaneous and uterine smooth muscle tumours is typically bland. Although leiomyosarcomas have been described in patients with FH mutations, this clinical association is rare and appears predominantly confined to people of Finnish origin.9 In addition to renal tumours with distinctive HLRCC papillary morphology, collecting duct carcinomas have also been described in association with HLRCC.10
More than 120 families have been reported with presumed pathogenic mutations in the FH gene, with mutations being predominantly missense, nonsense and frameshift changes. No correlation between genotype and phenotype has been reported: mutations that result in reduced FH activity are anticipated to produce a similar phenotype irrespective of mutation class.6 Two whole-gene deletions, which were 2.4 Mb and 1.9 Mb in size, were identified in one cohort of patients, although no phenotype data was given.5 One other whole-gene deletion was recently reported, with three affected family members having a combination of cutaneous and uterine leiomyomata, but no renal tumours.11
In-vitro studies suggest that FH-mutated renal tumours and von Hippel–Lindau (VHL) renal tumours share a common tumorigenic pathway, via dysregulation of hypoxia-inducible factor (HIF).12 The VHL protein couples changes in oxygen availability to gene expression through the regulation of HIF. Inactivation of the VHL gene results in increased HIF activity which, through increased expression of vascular endothelial growth factor (VEGF), platelet-derived growth factor ß (PDGFß) and transforming growth factor α (TGFα), plays a critical role in tumorigenesis. Because of the similar downstream effects of FH and VHL mutations, these factors represent rational therapeutic targets in HLRCC.
Sunitinib is a small-molecule multitargeted tyrosine kinase inhibitor that acts on VEGF and PDGFß in the context of renal cancers, while bevacizumab is a humanised monoclonal antibody targeting VEGF. Everolimus inhibits the mammalian target of rapamycin (mTOR), which is involved in HIF regulation.13 The use of these targeted therapies in advanced renal cell carcinoma has resulted in significant gains in progression-free survival compared with cytokine therapy or placebo, providing proof of principle for exploiting HIF-dependent pathways therapeutically. Inhibition of the epidermal growth factor receptor, a receptor tyrosine kinase through which TGFα acts, is being explored as a means to overcome TGFα overexpression. Both antibodies and small-molecule inhibitors, such as erlotinib, are under investigation.14
In summary, this case highlights the importance of recognising the unique pathological and clinical features of hereditary renal cancer. Not only does this facilitate early detection and genetic counselling in family members, in this age of personalised medicine such recognition may also serve to guide therapy.
Anatomical and histological features of the patient's renal cancer, and a photograph showing his skin rash
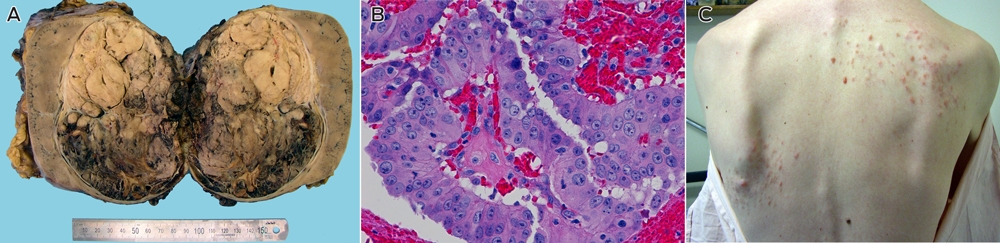
A: A 14 cm tumour in the renal medulla. B: Haematoxylin and eosin stained section of the renal tumour showing cells organised in papillary fronds with abundant eosinophilic cytoplasm, large nuclei and prominent inclusion-like eosinophilic nucleoli. C: The patient's skin rash, which was confirmed on biopsy to represent multiple cutaneous leiomyomata.
Competing interests
No relevant disclosures.
References
- Merino MJ, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 2007; 31: 1578-1585. _ENREF_1
- Gill AJ, Pachter NS, Clarkson A, et al. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. N Engl J Med 2011; 364: 885-886. _ENREF_2
- Kloepfer HW, Krafchuk J, Derbes V, Burks J. Hereditary multiple leiomyoma of the skin. Am J Hum Genet 1958; 10: 48-52. _ENREF_3
- Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 2001; 98: 3387-3392. _ENREF_4
- Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002; 30: 406-410. _ENREF_5
- Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 2008; 9: 20. _ENREF_6
- Pithukpakorn M, Wei MH, Toure O, et al. Fumarate hydratase enzyme activity in lymphoblastoid cells and fibroblasts of individuals in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 2006; 43: 755-762. _ENREF_7
- Wei MH, Toure O, Glenn GM, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 2006; 43: 18-27. _ENREF_8
- Ylisaukko-oja SK, Kiuru M, Lehtonen HJ, et al. Analysis of fumarate hydratase mutations in a population-based series of early onset uterine leiomyosarcoma patients. Int J Cancer 2006; 119: 283-287. _ENREF_9
- Kiuru M, Launonen V. Hereditary leiomyomatosis and renal cell cancer (HLRCC). Curr Mol Med 2004; 4: 869-875. _ENREF_10
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 2011; 79: 49-59. _ENREF_11
- Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 2005; 8: 143-153. _ENREF_12
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008; 372: 449-456. _Ref389663276
- Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011; 378: 1931-1939. _ENREF_14
Provenance: Not commissioned; externally peer reviewed.