





Volume 201 - Issue 1
Ipilimumab in pretreated patients with unresectable or metastatic cutaneous, uveal and mucosal melanoma
Authors: Marliese Alexander, James D Mellor, Grant McArthur and Damien Kee
Med J Aust 2014; 201 (1): 49-53. || doi: 10.5694/mja13.10448
Published online: 7 July 2014
Published online: 7 July 2014
The frequency and severity of adverse effects warrant experienced clinical supervision
Abstract
Objectives: To evaluate the efficacy and tolerability of ipilimumab in an Australian clinical setting, and to assess the association of response with melanoma subtype, BRAF mutation status, absolute lymphocyte count and incidence of serious immune-related adverse events (AEs).
Design, setting and participants: Retrospective review of patients with unresectable or metastatic melanoma treated with ipilimumab at an Australian oncology centre between July 2010 and April 2012.
Main outcome measures: Overall survival (OS), progression-free survival (PFS), incidence and severity of AEs.
Results: 104 patients were retrospectively followed for a median of 7 months (range 0–30 months). Median OS was 9.6 months (95% CI, 6.6–12.4), and median PFS was 3.0 months (95% CI, 2.7–3.4). The 1- and 2-year survival rates were 42% (95% CI, 32%–52%) and 18% (95% CI, 9%–30%), respectively. Median OS for patients with non-cutaneous (mucosal and uveal) melanomas was almost half that of patients with cutaneous melanoma: 5.8 months (95% CI, 2.8–12.4) v 11.7 months (95% CI, 7.1–13.8); P = 0.11. Raised absolute lymphocyte count was associated with increased PFS (P ≤ 0.005 at all measured time points) but not with OS (P > 0.15). Sex, age, brain metastases, BRAF mutation status, incidence of severe immune-related AEs and baseline lactate dehydrogenase levels did not affect OS or PFS (P > 0.05). Eighteen of 104 patients experienced serious AEs (≥ grade 3), including two treatment-related deaths.
Conclusion: In an Australian clinical practice setting, ipilimumab achieved efficacy and tolerability measures similar to those reported in clinical trials. The frequency and severity of ipilimumab-related AEs (including death) are notable, and treatment should occur under the supervision of an experienced clinical team.
Australia has the highest incidence of melanoma in the world; melanoma is the fourth most common form of cancer in Australia and the most common cancer in young Australians aged 15–39 years.1 While surgery remains the mainstay of treatment for early stage melanomas, there have been limited treatment options for more advanced disease and, historically, 5-year survival was less than 15%.2
Ipilimumab (Yervoy, Bristol-Myers Squibb) is a fully humanised monoclonal antibody directed against the cytotoxic T lymphocyte antigen 4 and the first treatment to demonstrate a survival benefit in advanced and metastatic disease (see Appendix 1 for a summary of published data). Pivotal studies of ipilimumab at 3 mg/kg in relapsed disease and 10 mg/kg in untreated disease demonstrated significant overall survival (OS) advantage compared with comparative arms (ipilimumab 3 mg/kg v vaccine: median OS, 10.1 v 6.4 months; hazard ratio for death, 0.66; P = 0.003;3 and 10 mg/kg plus dacarbazine v dacarbazine alone: median OS, 11.2 v 9.1 months; hazard ratio for death, 0.72; P < 0.0014). Ipilimumab has demonstrated clinical activity independent of negative prognostic markers (age, M stage or response to prior systemic therapy), offering a novel treatment to patients with poor prognosis.5 Immune-related adverse events (AEs) are frequently reported in clinical trials with serious events (grade ≥ 3) reported in 10%–56% of patients.3,4,6,7 However, with growing experience, the management of immune-related AEs has improved considerably and established protocols are widely available.8 Incidence of immune-related AEs and changes in lymphocyte count, presumably reflecting an immune response to therapy, have been reported to correlate with improved responses and increased survival.7,9,10
Ipilimumab was registered in Australia by the Therapeutic Goods Administration (TGA) in July 2011 for the treatment (monotherapy at 3 mg/kg) of unresectable or metastatic melanoma in patients who have progressed through or are intolerant to prior therapy.11 The Pharmaceutical Benefits Advisory Committee (PBAC) exhibited caution in recommending ipilimumab for inclusion in the Pharmaceutical Benefits Scheme (PBS) due to uncertainty surrounding clinical benefit and cost-effectiveness in the clinical setting.12 The PBAC public summary document stated that with limited sensitivity analyses conducted on survival data, a small change in the magnitude of any predicted benefit may significantly change the incremental benefit and cost-effectiveness of this already expensive agent; base-case incremental cost per quality-adjusted life-year gained is estimated to be between $45 000–$75 000.12 Thus, the subsequent positive recommendation for PBS listing requires ongoing data collection to determine whether the survival benefit modelled in the PBAC submission (and therefore the cost-effectiveness) can be realised in Australian clinical practice.
Of available published data (see Appendix 1), all except two studies7,9 are from the clinical trial setting. Supportive data from clinical practice settings, and more specifically from within Australia, are lacking. Our primary aim was to provide survival and toxicity outcomes data for Australian melanoma patients treated with ipilimumab in an Australian clinical practice setting. Secondary aims included the investigation of potential relationships between response and melanoma subtype, BRAF mutation status, absolute lymphocyte count (ALC) and immune-related AE.
Methods
Study design
A retrospective review of all patient records over specified time. Ethics approval was obtained from the Peter MacCallum Cancer Centre Divisional Review Panel and the study was conducted according to the National Health and Medical Research Council National Statement on Ethical Conduct in Human Research (http://www.nhmrc.gov.au/guidelines/publications/e72).
Patient population
Patients who commenced ipilimumab therapy between July 2010 (first patient treatment) and April 2012 were identified from hospital pharmacy dispensing records. Patients who received at least one dose of ipilimumab were included in the analysis. Patients were enrolled in an access program that enabled access to ipilimumab before TGA registration for patients, with the following eligibility criteria: a histologically confirmed diagnosis of unresectable (stage III) or metastatic (stage IV) cutaneous, uveal or mucosal melanoma; failure or intolerance to at least one prior treatment; aged ≥ 16 years; and Eastern Cooperative Oncology Group (ECOG) performance status of ≤ 2. According to local protocol, patients received ipilimumab at a dose of 3 mg/kg every 3 weeks for a total of four doses with reinduction offered to patients whose disease had progressed following either stable disease of ≥ 3 months duration or an initial objective response.
Data collection
The medical records of eligible patients were audited to retrieve the following patient- and disease-related factors: melanoma subtype and stage, presence of brain metastasis, lactate dehydrogenase (LDH), BRAF mutation status in exon 15 (BRAF-MT: mutant; BRAF-WT: wild type), past treatments, ECOG performance status, age and sex. Treatment-related information including AEs (serious AE defined as ≥ grade 3), ALC (high ALC defined as > 1 × 109/L), date of progression, and reason for cessation of ipilimumab (progressive disease, AE, death) were also collected. Patients were routinely reviewed every 3 weeks during treatment and every 4–6 weeks during surveillance. Radiological imaging was generally performed 6–8 weeks after induction and at least 8-weekly thereafter; however, a recognised limitation of the retrospective study design was an inability to ensure consistent radiological intervals. At each review, clinicians documented tolerability with standard reference to the United States Common terminology criteria for adverse events.13
Data analysis
Data were analysed using Stata version 12.0 (StataCorp). Survival end points were defined from the time of first ipilimumab dose until death in the case of overall survival; or on retrospective review of radiology reports or clinical notes suggesting progression or death, in the case of progression-free survival (PFS). For patients eligible for reinduction, a second PFS was calculated from the date of first reinduction dose of ipilimumab. Survival curves for OS and PFS were generated using the Kaplan–Meier method and compared using the log-rank test. For all statistical tests, a threshold of P < 0.05 was used to define significance.
Results
Patients and treatment
One hundred and four patients accessed ipilimumab via the named-patient program from July 2010 to April 2012. Patient characteristics are described in Appendix 2. Patients with uveal melanoma typically had more advanced disease, with stage M1c disease present in 100% of patients (compared with 84% of patients with cutaneous and 62% of patients with mucosal melanoma) and elevated LDH in 82% of patients (compared with 41% of patients with cutaneous and 25% of patients with mucosal melanoma). Patients were followed for a median of 7 months (range, 0–30 months). Seventy-three (70%) patients completed induction (four doses), with seven (7%) patients not completing treatment due to AEs and 24 (23%) due to progressive disease. Eighteen patients (17%) received other treatments for melanoma after ipilimumab.
Efficacy
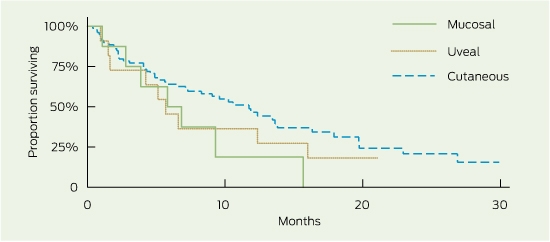
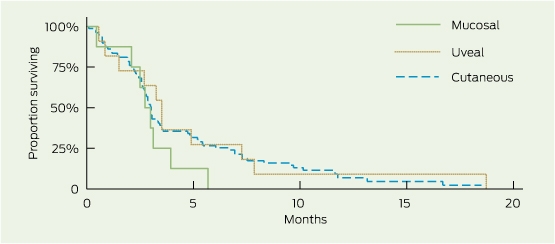
Median OS was 9.6 months (95% CI, 6.6–12.4), and median PFS was 3.0 months (95% CI, 2.7–3.4) (Box 1). The 1- and 2-year survival rates were 42% (95% CI, 32%–52%) and 18% (95% CI, 9%–30%), respectively. There were no non-melanoma-related deaths. Subgroup survival analysis is presented in Box 1. OS was similar for patients with and without brain metastasis (median OS, 9.3 and 10.6 months, respectively; log-rank test, P = 0.91), and PFS following reinduction therapy was significantly greater than PFS for the population as a whole (3.6 months: 95% CI, 3.6–undefined [upper limit not yet reached for patients receiving ≥ 2 reinduction ipilimumab doses] v 3 months: 95% CI, 2.7–3.4; P = 0.002). Patients with mucosal and uveal melanoma had reduced OS compared with those with cutaneous melanoma (5.8, 5.7 and 11.7 months, respectively), although this was not a statistically significant finding, likely due to small subset numbers (Box 2). PFS was similar across all groups (Box 2). Despite apparent numerical differences in OS by BRAF mutation status and incidence of serious immune-related AEs, small patient numbers resulted in wide confidence intervals and significance was not demonstrated. High ALC measured before or during ipilimumab induction was associated with an increased PFS (P ≤ 0.005) and numerically superior OS (P > 0.15).
Adverse events
Eighteen (17%) patients experienced a serious AE, with one patient experiencing two serious AEs (Box 1 and Box 3). Seven patients (7%) failed to complete induction due to an AE: diarrhoea/colitis (2), pruritus/rash (2), pituitary failure (1), myasthenia gravis (1) and hepatitis (1). Two patients died due to a treatment-related AE: autoimmune hypophysitis following the fourth dose of ipilimumab (1), and severe diarrhoea/colitis complicated by sepsis following the third dose of ipilimumab.
Discussion
Median OS (9.6 months) and 1-year OS (42%) exceeded proposed benchmark survival targets of 6.5 months for median OS and 25% for 1-year OS from a previous meta-analysis of second-line melanoma treatments.14 Survival outcomes were similar to those reported in the MDX010-20 study (median OS, 10.1 months)3 and in ipilimumab access schemes using a 10 mg/kg dosing regimen (median OS, 7.2–9 months).7,9 Median PFS (3.0 months) was also similar to the 2.9 months and 2.6–4.3 months found in the MDX010-20 and access scheme studies, respectively.3,7,9 With just under half of the study population (44/104) having brain metastasis at baseline, there was no difference in OS compared with patients without brain metastasis, suggesting that ipilimumab is an effective treatment option in both clinical situations. Encouragingly, PFS for patients undergoing reinduction therapy was similar to that seen following induction therapy, affording patients an efficacious subsequent treatment option and correlating with findings from recently published work extending on the MDX010-20 study.15 Competing clinical trials resulting in selection bias with respect to BRAF mutation status resulted in a study population that was not representative of the general melanoma population (BRAF-MT, 12% v 46%, respectively).16 Further, BRAF-MT patients in the study population were those likely to be ineligible for clinical trials or who had progressed following BRAF-inhibitor therapy (9/12 patients). This may be reflected in the poorer survival observed in this patient subgroup.
Although statistical significance is limited by the small number of patients with uveal (n = 11) and mucosal (n = 8) melanomas, there were some observed differences in survival data (Box 1) compared with the more prevalent cutaneous melanoma (n = 79). Trends demonstrated that future investigation of the shorter survival seen in the non-cutaneous subgroups may be warranted. The similar PFS but lower OS may reflect the different behaviour of the disease or the lack of subsequent treatment options compared with patients with cutaneous melanoma. The latter is unlikely in our patient cohort, where most patients (> 80%) with progressive disease following ipilimumab treatment were palliated (regardless of melanoma subtype) and not receiving subsequent therapy. Other explanatory factors may include the greater proportion of patients with uveal melanoma with widespread disease (M1c, 100%), and greater baseline tumour load (elevated LDH, 82%).
Raised ALC was highly correlated with prolonged PFS; however, although a trend was observed, the association with improved OS as shown in previous studies was not evident.7,9 There was a lower incidence of any immune-related AEs in our study (53/104, 51%) compared with the MDX010-20 study (80/131, 61%).3 However, this is likely to reflect poor documentation of low-grade AEs in the routine clinical setting compared with the highly regulated clinical trial setting with rigorous reporting requirements. When considering only serious immune-related AEs, assumed to be more accurately documented, incidence was more closely aligned: 14% (15/104) of patients in the present study compared with 15% (19/131) in the MDX010-20 study. We report similar incidence of overall skin immune-related AEs (41/104, 39% v 57/131, 44%), grade 3–4 skin immune-related AEs (3/104, 3% v 3/131, 2%) and grade 3–4 diarrhoea/colitis (8/104, 8% v 13/131, 10%). The incidence of treatment-related deaths (grade 5 AE) was comparable, with 2% (2/104) and 3% (4/131) treatment-related mortality rates reported in the current and MDX010-20 studies, respectively.
We reported the clinical details pertaining to the rare and serious ipilimumab AEs experienced by our patients, including two grade 5 events. One patient who had been successfully managed with high-dose steroids for grade 4 diarrhoea/colitis underwent surgical excision of brain metastases. The patient developed colonic microperforation and sepsis, and died, after the steroid dose was tapered postoperatively. The problem of rebound symptoms is well recognised and, consequently, guidelines recommend a prolonged steroid taper over 45–60 days.8 This case highlights the importance of proper communication to patients and between treating teams. Two patients experienced serious autoimmune hypophysitis, with one subsequent death. The death was a result of delayed recognition of symptoms with an initial working diagnosis of lithium toxicity delaying steroid administration. In the second patient, symptoms developed 1 month after induction therapy. Earlier recognition and treatment with oral steroids and hormone replacement resulted in both a symptomatic and biochemical response, and the patient has maintained a sustained melanoma response to therapy. A single case of grade 3 pituitary failure was attributed to disease progression when magnetic resonance imaging scans unexpectedly demonstrated the presence of extensive bone metastases involving the pituitary fossa. One patient experienced myasthenia gravis, a rare (< 1% incidence) immune-related AE associated with ipilimumab. The first two doses were complicated by sweats and lethargy; following the third dose, the patient presented with dysarthria, dysphasia, dysphonia and diplopia. Myasthenia gravis diagnosis was confirmed by elevated levels of antiacetylcholinesterase antibody. Ipilimumab was ceased and symptomatic improvement was achieved using steroids, pyridostigmine and intravenous immunoglobulin-γ (unpublished case report). The frequency and severity of ipilimumab-related AEs (including death), although not dissimilar to rates reported in the clinical trial setting, are notable and warrant the recommendation that ipilimumab treatment should occur only under the supervision of an experienced clinical team.
1 Overall survival (OS) and progression-free survival (PFS), by subgroup
No. of patients | Median OS, months | 95% CI | P | Median PFS, months | 95% CI | P | |||||||||
All patients | 104 | 9.6 | 6.6–12.4 | 3.0 | 2.7–3.4 | ||||||||||
Melanoma subtype* | |||||||||||||||
Cutaneous | 79 | 11.7 | 7.1–13.8 | 0.11 | 3.0 | 2.7–3.4 | 0.72 | ||||||||
Non-cutaneous | 19 | 5.8 | 2.8–12.4 | 3.1 | 2.1–3.9 | ||||||||||
Uveal | 11 | 5.7 | 1.5–16.0 | 3.5 | 0.9–7.33 | ||||||||||
Mucosal | 8 | 5.8 | 1.1–nd | 2.7 | 0.5–3.9 | ||||||||||
Sex | |||||||||||||||
Male | 71 | 9.6 | 5.8–13.4 | 0.90 | 3.0 | 2.6–3.5 | 0.69 | ||||||||
Female | 33 | 7.5 | 5.1–15.7 | 3.0 | 2.7–3.5 | ||||||||||
Age | |||||||||||||||
≤ 65 years | 57 | 10.9 | 7.1–13.6 | 0.23 | 3.1 | 2.7–3.9 | 0.83 | ||||||||
> 65 years | 47 | 6.7 | 3.9–16.3 | 2.7 | 2.4–3.4 | ||||||||||
Brain metastases | |||||||||||||||
Yes | 44 | 9.3 | 4.9–12.4 | 0.91 | 3.0 | 2.8–3.9 | 0.32 | ||||||||
No | 60 | 10.6 | 5.6–15.7 | 2.7 | 2.2–3.5 | ||||||||||
BRAF mutation* | |||||||||||||||
Positive | 12 | 4.9 | 1.0–12.4 | 0.11 | 2.9 | 0.4–6.6 | 0.53 | ||||||||
Negative | 80 | 11.7 | 7.5–16.3 | 3.0 | 2.7–3.6 | ||||||||||
Baseline LDH level > 460 U/L | |||||||||||||||
Yes | 56 | 6.6 | 4.2–13.6 | 0.27 | 2.9 | 2.4–3.4 | 0.31 | ||||||||
No | 43 | 11.7 | 7.3–15.7 | 3.1 | 2.7–5.7 | ||||||||||
irAE ≥ grade 3 | |||||||||||||||
Yes | 17 | 13.6 | 5.3–nd | 0.41 | 2.9 | 2.4–5.5 | 0.95 | ||||||||
No | 87 | 8.3 | 5.7–12.4 | 3.0 | 2.7–3.5 | ||||||||||
ALC > 1 × 109/L* | |||||||||||||||
Baseline† | |||||||||||||||
Yes | 65 | 10.9 | 5.8–13.8 | 0.23 | 3.5 | 2.9–4.9 | 0.002 | ||||||||
No | 35 | 7.1 | 2.6–11.9 | 2.6 | 2.0–3.0 | ||||||||||
After ipilimumab dose 1‡ | |||||||||||||||
Yes | 70 | 12.4 | 9.3–16.3 | 0.15 | 3.5 | 3.0–5.3 | < 0.001 | ||||||||
No | 17 | 6.8 | 3.8–19.7 | 2.6 | 2.0–3.1 | ||||||||||
After ipilimumab dose 2‡ | |||||||||||||||
Yes | 65 | 12.4 | 9.6–17.9 | 0.23 | 3.5 | 3.0–5.3 | 0.005 | ||||||||
No | 14 | 6.8 | 2.3–nd | 2.6 | 2.0–3.3 | ||||||||||
ALC = absolute lymphocyte count. CNS = central nervous system. irAE = immune-related adverse event. LDH = lactate dehydrogenase. nd = upper limit of CI not defined (not yet reached for patients receiving ≥ 2 reinduction ipilimumab doses). * Patients with missing or unknown data excluded from analysis. † Before receiving ipilimumab. ‡ Of four planned ipilimumab doses. | |||||||||||||||
2 Overall survival and progression-free survival, by melanoma subtype*
Overall survival
Progression-free survival
* Overall log-rank test for equality: overall survival, P = 0.204; progression-free survival; P = 0.397.
3 Number of patients experiencing adverse events, by grade* (n = 104)
No. of patients with adverse events | |||||||||||||||
Adverse event | Total | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | |||||||||
Any† | 70 | 58 | 14 | 15 | 2 | 2 | |||||||||
Non-immune-related | |||||||||||||||
Any† | 41 | 34 | 7 | 4 | – | 1 | |||||||||
Nausea | 20 | 15 | 3 | 2 | – | – | |||||||||
Vomiting | 8 | 4 | 2 | 2 | – | – | |||||||||
Headache | 4 | 2 | 2 | – | – | – | |||||||||
Fatigue | 23 | 20 | 2 | 1 | – | – | |||||||||
Abdominal pain | 7 | 6 | – | 1 | – | – | |||||||||
Uveal irritation | 4 | 2 | 2 | – | – | – | |||||||||
Infection | 4 | 1 | 2 | – | – | 1 | |||||||||
Immune-related | |||||||||||||||
Any† | 53 | 48 | 7 | 12 | 2 | 1 | |||||||||
Gastrointestinal | 21 | 13 | – | 7 | 1 | – | |||||||||
Colitis | 8 | 2 | – | 5 | 1 | – | |||||||||
Diarrhoea | 19 | 12 | – | 6 | 1 | – | |||||||||
Hepatic | 3 | 2 | – | 1 | – | – | |||||||||
Hepatitis | 1 | – | – | 1 | – | – | |||||||||
Elevated LFT results‡ | 3 | 2 | – | 1 | – | – | |||||||||
Endocrine | 8 | 3 | 2 | 2 | 1 | ||||||||||
Hypophysitis | 2 | – | – | 1 | – | 1 | |||||||||
Hypopituitarism | 6 | 3 | 2 | 1 | – | – | |||||||||
Skin† | 41 | 35 | 5 | 3 | – | – | |||||||||
Pruritus | 30 | 24 | 3 | 3 | – | – | |||||||||
Rash | 34 | 28 | 3 | 3 | – | – | |||||||||
Other | 4 | 3 | 1 | – | – | – | |||||||||
Myasthenia gravis | 1 | – | – | – | 1 | – | |||||||||
LFT = liver function test.* According to the Common terminology criteria for adverse events.13 † Patients who experienced separate events of different grades and/or of different types were counted separately under each grade and/or type, but only once in the total count. ‡ Any one or more of alkaline phosphatase: reference interval (RI), 30–120 U/L; alanine aminotransferase: RI, 0–50 U/L; bilirubin: RI, 0–17 µmol/L. | |||||||||||||||
Competing interests
Damien Kee has previously received honoraria from Bristol-Myers Squibb for educational presentations. This article has been prepared completely independently of any input from Bristol-Myers Squibb.
References
- Australian Institute of Health and Welfare, Australasian Association of Cancer Registries. Cancer in Australia: an overview, 2012. Canberra: AIHW, 2012. (AIHW Cat. No. CAN 70; Cancer Series No. 74.) http://www.aihw.gov.au/publication-detail/?id=60129542359 (accessed Nov 2013).
- Balch CM, Soong SJ, Gershenwald JE, et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol 2001; 19: 3622-3634. 2
- Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711-723. 3
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364: 2517-2526. 4
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010; 11: 155-164. 5
- Hersh EM, O'Day SJ, Powderly J, et al. A phase II multicenter study of ipilimumab with or without dacarbazine in chemotherapy-naive patients with advanced melanoma. Invest New Drugs 2011; 29: 489-498. 6
- Di Giacomo AM, Danielli R, Calabrò L, et al. Ipilimumab experience in heavily pretreated patients with melanoma in an expanded access program at the University Hospital of Siena (Italy). Cancer Immunol Immunother 2011; 60: 467-477. 7
- Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 2012; 30: 2691-2697. 8
- Ku GY, Yuan J, Page DB, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer 2010; 116: 1767-1775. 9
- Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 2012; 13: 459-465. 10
- Therapeutic Goods Administration. Australian Public Assessment Report for ipilimumab. August 2011. http://www.tga.gov.au/pdf/auspar/auspar-yervoy.pdf (accessed Oct 2013).
- Pharmaceutical Benefits Advisory Committee. Ipilimumab (Yervoy). Public summary document. March 2012. http://www.pbs.gov.au/industry/listing/elements/pbac-meetings/psd/2012-03/ipilimumab.pdf (accessed Oct 2013).
- US Department of Health and Human Services National Institutes of Health, National Cancer Institute. Common terminology criteria for adverse events. Version 4.0. May 2009. http://www.acrin.org/Portals/0/Administration/Regulatory/CTCAE_4.02_2009-09-15_QuickReference_5x7.pdf (accessed Oct 2013).
- Korn EL, Liu PY, Lee SJ, et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J Clin Oncol 2008; 26: 527-534. 14
- Robert C, Schadendorf D, Messina M, et al; MDX010-20 investigators. Efficacy and safety of retreatment with ipilimumab in patients with pretreated advanced melanoma who progressed after initially achieving disease control. Clin Cancer Res 2013; 19: 2232-2239. 15
- Menzies AM, Haydu LE, Visintin L, et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res 2012; 18: 3242-3249. lefthere