




Necrobiotic xanthogranuloma is a rare, multisystem granulomatous disease that can have skin and visceral organ involvement. It is strongly associated with paraproteinaemia, and has rare potential for transformation to myeloma or lymphoma, warranting ongoing surveillance. We report a challenging case of necrobiotic xanthogranuloma, believed to be the first reported case of hepatic involvement.
Clinical record
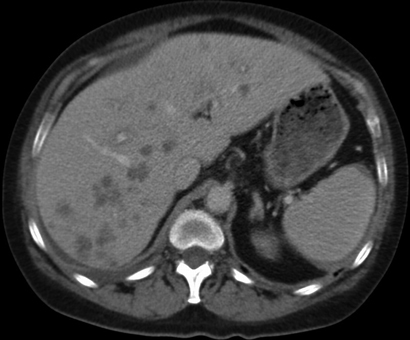
A 51-year-old woman was referred to hospital after an incidental finding of abnormal liver function test results (γ-glutamyl transpeptidase, 271 U/L [reference interval (RI), < 60 U/L]; alkaline phosphatase, 220 U/L [RI, 30–110 U/L]; hepatic transaminases, normal), acute renal impairment (urea, 11.7 mmol/L [RI, 2.7–8.0 mmol/L]; creatinine, 207 µmol/L [RI, 50–100 µmol/L]) and elevated globulin levels of 44 g/L (RI, 21–41 g/L). She was otherwise well and asymptomatic. Her past medical history included resistant hypertension with renal denervation 8 months earlier, and chronic macrocytic anaemia secondary to alcohol. Physical examination was largely unremarkable apart from several skin nodules and hepatomegaly (4 cm below the costal margin). She underwent an abdominal ultrasound, which showed multiple hypoechoic liver lesions suspicious for metastasis. An abdominal computed tomography (CT) scan (Box 1) confirmed the presence of hypovascular liver lesions suggestive of metastasis. Malignancy screening, including mammography, colonoscopy and CT of the chest, abdomen and pelvis, was negative. A subsequent liver biopsy specimen showed necrotising granulomas with no evidence of malignancy. The initial histopathological impression was of mycobacterial infection, despite negative Ziehl–Neelsen and Gram stains and the absence of infective organisms.
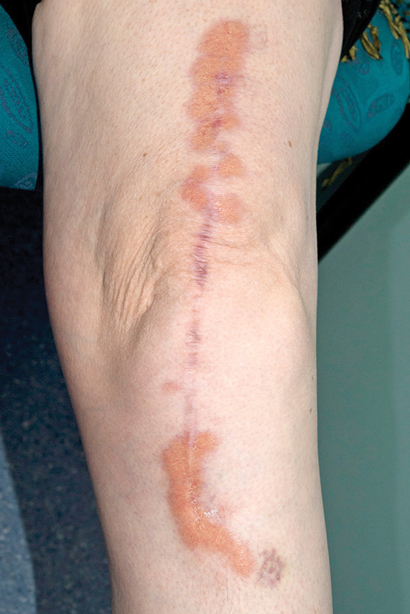
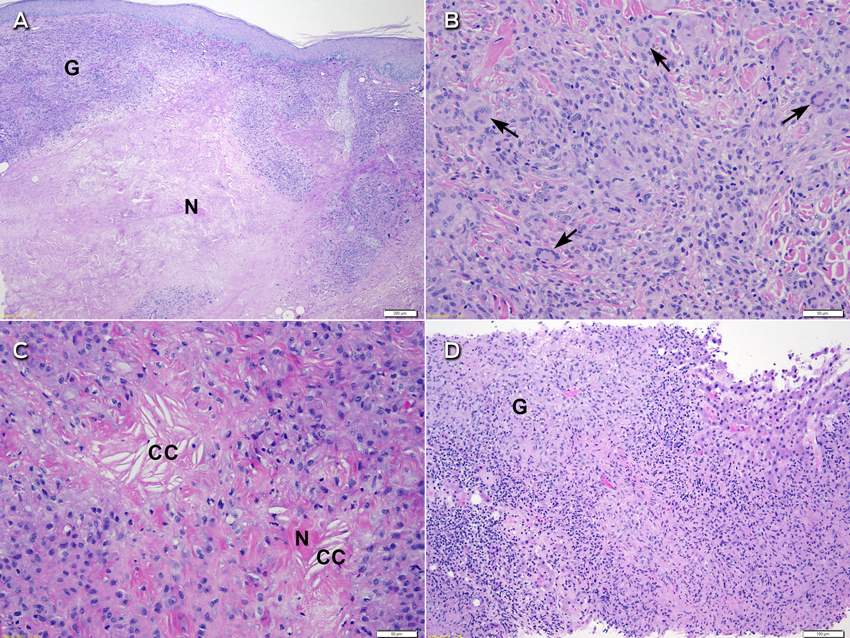
Further assessment found no risk factors for tuberculosis exposure and no symptoms of underlying infection. Dermatology consultation was requested and the skin nodules were closely examined. These included a 1 × 2 cm violaceous nodule on the left side of the woman's neck, a 2 × 5 cm yellowish plaque on her left flank, and a large violaceous-yellow plaque exhibiting Koebner phenomenon along the scar from her left total knee replacement surgery (Box 2). The patient reported that they had been present for 8–12 months and were asymptomatic. Biopsy specimens from the lesions on her neck and flank showed identical histological features to the liver specimen, again negative for Mycobacterium tuberculosis on Ziehl–Neelsen stain, culture and polymerase chain reaction (PCR) testing. A previous biopsy sample of the plaque overlying her knee-replacement scar had been taken by a dermatologist and showed non-discrete granulomatous inflammation and necrobiosis (Box 3, A). In retrospect, this was consistent with her current skin sample.
Thorough investigations were performed for causes of systemic granulomatous diseases. Infective causes that were investigated included histoplasmosis, brucellosis and mycobacterial infections. However, results from extensive serological testing, cultures, tuberculosis PCR testing of the skin and liver biopsy samples, and an interferon-γ release assay were negative. Non-infective differential diagnoses included sarcoidosis or vasculitis. However, angiotensin-converting enzyme concentration was low and no hilar lymphadenopathy was found on CT of the chest. Results of autoimmune and vasculitic screening were unremarkable except for low-titre antinuclear antibody (ANA) (titre, 1 : 80; pattern, speckled) and type 1 monoclonal cryoglobulinaemia (1.2 g/L). Elevation of the patient's erythrocyte sedimentation rate (ESR) was also noted (> 120 mm/h [RI, 1–20 mm/h]). The patient was assessed for multiple myeloma, given that she had anaemia (haemoglobin, 106 g/L [RI, 115–155 g/L]), renal impairment, elevated globulin and hypercalcaemia (corrected calcium, 2.72 mmol/L [RI, 2.10–2.55 mmol/L]). She was found to have paraproteinaemia (11 g/L), with raised levels of immunoglobulin G (IgG) kappa light chains (36 mg/L [RI, 3–19 mg/L]) and β2-microglobulin (6.9 mg/L [RI, 0.6–2.9 mg/L]), and positive urine Bence Jones protein of 0.01 g/L. Urine sediment was otherwise bland. A skeletal survey was normal and a bone marrow biopsy sample showed no evidence of plasmacytosis or a lymphoproliferative disorder (plasma cells, 3%). The patient was diagnosed with monoclonal gammopathy of undetermined significance (MGUS).
The biopsy specimens were reviewed for a second time for consideration of a non-infective aetiology. On the skin specimen (Box 3, B and C), in addition to scattered Langhans-type giant cells, several of the giant cells were more characteristic of Touton-type giant cells with a peripheral foamy cytoplasm. While necrosis was prominent, there were foci of necrobiosis with needle-shaped cholesterol clefts. Given the patient's clinical history, associated MGUS and characteristic histological features in the absence of any detectable infective aetiology, the lesions were in keeping with the diagnosis of necrobiotic xanthogranuloma.
Discussion
Necrobiotic xanthogranuloma (NXG) is a rare, chronic and progressive systemic granulomatous disease first described by Kossard and Winkelmann in 1980.1 It manifests as destructive cutaneous lesions that may be cosmetically disfiguring. It is usually found in the periorbital area; however, other cutaneous sites such as the trunk or extremities may be affected, and involvement of scars has been reported.2 Interestingly, our patient did not have periorbital lesions. Involvement of systemic organs such as the respiratory tract, heart, spleen, kidney, intestine, ovary, lacrimal gland, parotid gland, bone marrow, skeletal muscles and brain has been widely recognised.3-5
Characteristic histopathological changes3 are paramount to diagnosis. They consist of palisading histiocytic xanthogranuloma with bands of hyaline necrobiosis (cell degeneration of the collagen bundles). Touton giant cells, atypical foreign-body giant cells and cholesterol clefts are often seen, while foam cells and lymphoid nodules may also be present. Other examples of necrobiotic palisading granulomas include granuloma annulare, necrobiosis lipoidica and rheumatoid nodules. These are distinct from tuberculoid granulomas, which are confluent with a rim of lymphocytes and, sometimes, caseation necrosis. Other granulomatous conditions include discrete sarcoidal granulomas, suppurative granulomas and foreign-body granulomas — none of which should demonstrate necrobiosis or xanthomatous changes as seen in NXG.
Histological changes of NXG are prominent in the skin, but some features have been observed in visceral organs. Although associated hepatomegaly has been observed,2 histological examination of the liver has only been reported in a few cases — two patients showed reactive changes including nodular transformation,4,6 and one showed amyloidosis.7 Syncytial giant cell hepatitis was reported in a patient with NXG, but there was no more than a tenuous association.8 Our patient's liver biopsy specimen (Box 3, D) showed necrobiosis and necrosis associated with diffuse granulomatous infiltrate similar to that seen in the skin specimens. As NXG can lead to necrobiotic granulomatous inflammation in other organs, it is reasonable to expect that similar infiltrate may occur within the liver. We believe this is the first reported case of true liver involvement of NXG.
An important association in patients with NXG is the presence of paraproteinaemia, evident in 80%–90%.3 Our patient had IgG kappa, which is more common among patients with NXG and paraproteinaemia (60%) than IgG lambda (24%).3 Moreover, patients with NXG are at risk of developing haematological malignancies, with multiple myeloma occurring in 10%.9 Other reported associations include Hodgkin and non-Hodgkin lymphoma, chronic lymphocytic leukaemia, myelodysplastic syndrome, macroglobulinaemia, cryoglobulinaemia and amyloidosis.3 These haematological disorders may emerge before or after the onset of skin lesions (ranging from 8 years before to 11 years after).10 Surveillance is therefore crucial for early detection of malignancy. Other laboratory findings commonly seen in NXG include elevated ESR and positive ANA, which were also noted in our patient.3,9,11
Although the aetiology of NXG is unclear, several hypotheses have been proposed. Paraproteinaemia has been considered as a primary cause, whereby paraproteins act as autoantibodies and form complexes with lipids,5 or function as a lipoprotein and bind to the receptors of macrophages.12 However, one reported case of extension of cutaneous plaques despite successful reduction in paraproteinaemia calls into question its significance in the pathogenesis of NXG.13 Paraproteinaemia may be a secondary phenomenon rather than a primary cause. It has been proposed that activated monocytes with enhanced phagocytic ability can accumulate lipids and deposit them in the skin causing a giant-cell inflammatory response.14 More recently, an infectious process has also been proposed, based on the discovery of spirochaetes (Borrelia spp) in NXG lesions.15
Currently, no first-line recommended therapy for NXG has been established. Given the rarity of the condition, no randomised controlled trials have been conducted and treatment relies on anecdotal evidence. A variety of therapies have been described in the literature, most of which treat the skin lesions or associated haematological condition rather than visceral organ involvement. Treatment with corticosteroids, alkylating agents, antimetabolites, surgical excision and other agents have been described but have shown inconsistent results.3
The case we report here not only showed that NXG may involve changes in the liver, but illustrates the complexity of diagnosing systemic NXG. About 100 cases of NXG have been reported and only a few have included visceral involvement. It is therefore understandable that this rare condition did not feature on our initial differential list of systemic granulomatous diseases. Importantly, most reported cases have been discovered during autopsy,3-5 implying that signs and symptoms of systemic disease are not obvious. Diagnosis of our patient's systemic disease was made on the basis of the concomitant cutaneous plaques showing xanthomatous and granulomatous changes on histological examination and the associated IgG kappa paraproteinaemia. In her case, the liver involvement was more substantial than the skin nodules, which remained inconspicuous and were not cosmetically disfiguring. Her liver function tests have remained stable, as has her paraproteinaemia; therefore, our recommendation is for rigorous surveillance, with initiation of active treatment only if there is progression of paraproteinaemia.
The patient's renal impairment was unrelated to NXG; it was due to bilateral renal artery stenosis, a rare complication of renal denervation. She underwent bilateral renal artery stenting and her renal function improved.
In conclusion, NXG is a rare systemic granulomatous disorder that usually manifests as characteristic cutaneous plaques. It may involve a number of internal organs with or without associated paraproteinaemia and cryoglobulinaemia. These systemic features may assist in diagnosis when skin features are not characteristic and histological features are subtle. To our knowledge, this is the first reported case of NXG involving the liver. Given its strong association with paraproteinaemia, ongoing surveillance is recommended for potential development of haematological malignancies. There is no specific recommended therapy, and the decision to treat should be based on the individual patient.
- 1. Kossard S, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. J Am Acad Dermatol 1980; 3: 257-270.
- 2. Koch P, Goerdt S, Géraud C. Erythematous papules, plaques, and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol 2013; 149: 1103-1104.
- 3. Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol 2009; 48: 1-10.
- 4. Novak PM, Robbins TO, Winkelmann RK. Necrobiotic xanthogranuloma with myocardial lesions and nodular transformation of the liver. Hum Pathol 1992; 23: 195-196.
- 5. Bullock JD, Bartley GB, Campbell RJ, et al. Necrobiotic xanthogranuloma with paraproteinemia. Case report and a pathogenetic theory. Ophthalmology 1986; 93: 1233-1236.
- 6. Hunter L, Burry AF. Necrobiotic xanthogranuloma: a systemic disease with paraproteinemia. Pathology 1985; 17: 533-536.
- 7. Westermann G, August C, Bonsmann G, et al. [Necrobiotic xanthogranuloma with skin and liver amyloidosis] [German]. Med Klin (Munich) 2001; 96: 50-54.
- 8. Amer R, Pe'er J, Pappo O, Dotan S. Necrobiotic xanthogranuloma associated with choroidal infiltration and syncytial giant cell hepatitis. J Neuroophthalmol 2005; 25: 189-192.
- 9. Martínez Fernández M, Rodríguez Prieto MA, Ruiz González I, et al. Necrobiotic xanthogranuloma associated with myeloma. J Eur Acad Dermatol Venereol 2004; 18: 328-331.
- 10. Ugurlu S, Bartley GB, Gibson LE. Necrobiotic xanthogranuloma: long-term outcome of ocular and systemic involvement. Am J Ophthalmol 2000; 129: 651-657.
- 11. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol 1992; 128: 94-100.
- 12. Rappersberger K, Wrba F, Heinz R, et al. [Necrobiotic xanthogranuloma in paraproteinemia] [German]. Hautarzt 1989; 40: 358-363.
- 13. Ziemer M, Wedding U, Sander CS, Elsner P. Necrobiotic xanthogranuloma – rapid progression under treatment with melphalan. Eur J Dermatol 2005; 15: 363-365.
- 14. Matsuura F, Yamashita S, Hirano K, et al. Activation of monocytes in vivo causes intracellular accumulation of lipoprotein-derived lipids and marked hypocholesterolemia – a possible pathogenesis of necrobiotic xanthogranuloma. Atherosclerosis 1999; 142: 355-365.
- 15. Zelger B, Eisendle K, Mensing C, Zelger B. Detection of spirochetal micro-organisms by focus-floating microscopy in necrobiotic xanthogranuloma. J Am Acad Dermatol 2007; 57: 1026-1030.
No relevant disclosures.
* Equal first authors