





Volume 199 - Issue 8
Primary amenorrhoea with hypertension: undiagnosed 17-α-hydroxylase deficiency
Authors: Fiona L Britten, Kimberly B Ulett, Emma L Duncan and Donald A Perry-Keene
Med J Aust 2013; 199 (8): 556-558. || doi: 10.5694/mja12.11619
Published online: 21 October 2013
Published online: 21 October 2013
17-α-hydroxylase deficiency is a rare cause of congenital adrenal hyperplasia that often goes undiagnosed into adulthood. Clinicians’ awareness of the presenting characteristics that distinguish this deficiency from more common forms of the disease is important.
Clinical record
A 22-year-old Taiwanese woman on a working holiday in Australia was admitted to hospital in March 2011 with a cervical spine fracture from a motor vehicle accident. During her hospital stay it was noted that she had metabolic alkalosis with significant hypokalaemia.
Her past medical history included presentation at 16 years of age with primary amenorrhoea that was attributed to pubertal delay. At that time, she was given oestrogen–progesterone replacement therapy, which induced withdrawal bleeding. She noted an increase in stature after commencing hormone replacement therapy, growing to the height of her two brothers. She was diagnosed with mild hypertension at a pre-immigration health check in Taiwan. After arriving in Australia, 12 months before the accident, she had stopped taking hormonal therapy and experienced complete cessation of withdrawal bleeds. She was ethnically Han Chinese with no family history of consanguinity.
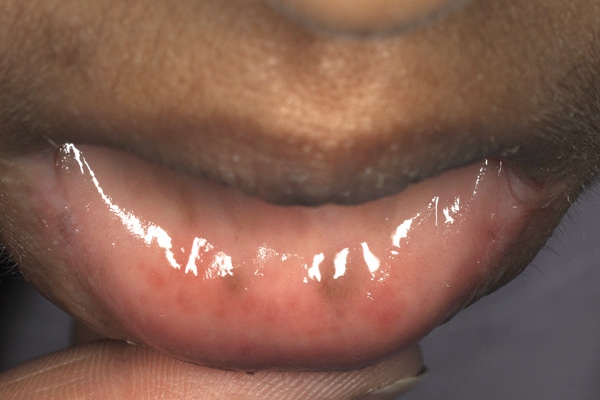
Physical examination revealed she was tall and thin, with height 174 cm and body mass index 19.6 kg/m2. Her blood pressure was elevated, with a maximal reading of 190/120 mmHg. In addition to bruising consistent with her injury, she had hyperpigmentation of the oral mucosa and axillary areas (Box 1). Pubic and axillary hair were absent, and breast development was minimal (Tanner stage 2). An abdominal computed tomography scan showed bulky adrenal glands and an atrophic uterus. Her potassium level was 2.4 mmol/L and an initial electrocardiogram showed sinus rhythm and hypokalaemic changes, including a prolonged corrected QT interval, but no left ventricular hypertrophy.
The medical team prescribed verapamil 80 mg three times daily. Potassium replacement in excess of 120 mmol/day was required to maintain a potassium level of 2.7–3.0 mmol/L. Advice was sought from endocrinologists.
On review, given the patient’s history, clinical signs and biochemistry profile, an upstream block in steroid synthesis was suspected. Further biochemical testing showed low levels of serum cortisol, oestradiol and testosterone and high levels of adrenocorticotrophic hormone, gonadotrophins and progesterone. Serum aldosterone and renin levels were low (Box 2). These results are consistent with 17-α-hydroxylase deficiency. This diagnosis was further supported by steroid metabolite levels in a spot urine sample. Pregnanediol (a progesterone metabolite), tetrahydrocorticosterone and tetrahydro compound A (deoxycorticosterone metabolites) were elevated. In addition, decreased 11-deoxycortisol and cortisol metabolites (tetrahydrodeoxycortisol, tetrahydrocortisone and tetrahydrocortisol) and decreased androstenedione metabolites (etiocholanolone and androsterone) were observed.
Sequencing of CYP17A1 demonstrated homozygosity for a non-synonymous mutation (ie, one that changes the amino acid sequence of the protein) previously described in the Chinese population. A karyotype analysis was not performed, owing to the patient’s history of withdrawal bleeds with hormone replacement therapy and visualisation of the uterus on imaging that was performed because of her traumatic injuries.
The patient was commenced on prednisolone 10 mg twice daily and spironolactone 25 mg daily. Hypokalaemia and hypertension resolved rapidly, and potassium supplementation, spironolactone and verapamil were no longer required. Recommencement of sex hormone replacement and weaning of prednisone to a physiological dose were planned for after her return to Taiwan.
Generally considered the diagnostic domain of the paediatrician, congenital adrenal hyperplasia (CAH) is most frequently associated with 21-hydroxylase deficiency and usually presents in neonates with salt wasting and/or ambiguous genitalia. In contrast, a less common form of CAH, 17-α-hydroxylase deficiency, may remain asymptomatic and undiagnosed into early adulthood. There have been over 150 cases reported in the literature, with one previous case report in Australia.1 We describe a case of 17-α-hydroxylase deficiency discovered incidentally in a patient admitted after trauma.
CAH is an autosomal recessive disorder that involves a defective enzyme step in the biosynthetic pathway of steroid hormones, most commonly a 21-hydroxylase deficiency. An absence of sufficient cortisol means there is no negative feedback at the level of the hypothalamus. This leads to increased adrenocorticotrophic hormone (ACTH) production that stimulates adrenal hyperplasia and the production of steroid precursor products upstream of the defective enzyme. A deficiency of 17-α-hydroxylase is an uncommon cause of CAH with a characteristic phenotype at odds with the clinical features of the most common types of CAH.
17-α-hydroxylase deficiency is due to mutations of CYP17A1, located on chromosome 10q24.3. It encodes the enzyme CYP17A1 which catalyses both 17-α-hydroxylase and 17,20-lyase activity, essential for production of cortisol, androgens and oestrogen. In a person with this deficiency, the steroid precursor products proximal to the enzymatic block are converted to progesterone and then to deoxycorticosterone (DOC) and corticosterone by way of the intact remaining enzyme pathway (Box 3). Aldosterone is usually suppressed, owing to the salt-retaining and volume expansion effects of the high concentrations of DOC, with consequent transcriptional downregulation of aldosterone synthase.2 In our patient, the serum aldosterone level initially appeared high when measured by radioimmunoassay, owing to interfering compounds; when the same sample was retested by high performance liquid chromatography, it was actually low. Follicle stimulating hormone (FSH) and luteinising hormone (LH) concentrations were high, as expected because of lack of negative feedback from sex steroids.
The true incidence of 17-α-hydroxylase deficiency is unknown but may be around 1 : 70 000.3 Most known mutations appear to be novel mutations, with 97 now characterised in the Human Gene Mutation Database.4 In several populations, however, common mutations unique to that ethnic group suggest a founder effect. These groups include the Canadian Mennonites and Dutch Frieslander,5 the Japanese,6 the Spanish–Portuguese in Brazil,7 Guamanians8 and the Chinese. Several common mutations unique to the Chinese population have been reported.9-12 A recent review of 26 Chinese families with 17-α-hydroxylase deficiency found 15 mutations overall, many unique to the Chinese population. Of these, 10 patients from six families carried a nine-base-pair deletion on exon 8 (D487-S488-F489); and 15 patients from 15 families carried a deletion-insertion mutation on exon 6 (Y329fs) — the mutation identified in our patient.13 The frequency of each mutation remains rare in the Han Chinese population, with a recent study showing three heterozygote carriers of the D487 mutation and six of the Y329fs mutation among 3245 healthy Han Chinese individuals.11
To date, there has been only one previous case report of 17-α-hydroxylase deficiency in Australia. This may reflect either non-publication of diagnosed cases or underdiagnosis. In a Brazilian cohort of patients in whom 17-α-hydroxylase deficiency was the second most common cause of CAH, 60% of patients were initially misdiagnosed.3 In one Chinese hospital, 17-α-hydroxylase deficiency was the second most commonly diagnosed cause of CAH over a 5-year period.10
The condition should be considered in young women who fail to enter puberty and have concomitant hypertension. With no exposure to testosterone in utero, patients with 17-α-hydroxylase deficiency have external female genitalia regardless of male (XY) or female (XX) karyotype. XX females have a uterus but do not develop secondary sexual characteristics such as pubic hair or breast tissue, nor do they undergo menarche. XY individuals do not have a uterus or fallopian tubes because the testes produce Müllerian inhibitory factor, resulting in Müllerian duct regression. Males with partial insufficiency can present with ambiguous external genitalia or male pseudohermaphroditism.14 Tall stature results from failure of epiphyseal fusion due to lack of oestrogen. DOC-mediated mineralocorticoid excess results in hypertension and hypokalaemia; the postural hypotension from cortisol and aldosterone insufficiency, seen in 21-hydroxylase deficiency, is absent. When elevated, corticosterone has sufficient activity at glucocorticoid receptors to prevent other significant effects of cortisol insufficiency such as hypoglycaemia. However, elevated ACTH levels may result in hyperpigmentation, as in our patient. The diagnosis is further established by the characteristic pattern of high levels of steroid precursors and their urinary metabolites (Box 2). Important aspects of a patient’s history include ethnicity, familial consanguinity, and pubertal development. Mutations of CYP17A1 can be confirmed by genetic testing, and a karyotype analysis is also usually performed.
Treatment consists, first, of physiological doses of glucocorticoids to replace cortisol and to suppress ACTH-driven mineralocorticoid (ie, DOC) excess, using the smallest dose possible to avoid glucocorticoid overreplacement. Dexamethasone (0.25 to 0.5 mg), prednisone (2–4 mg/m2) or hydrocortisone (10–15 mg/m2) may all be used.3 The dose is titrated to normalise blood pressure and potassium. Usually, the plasma renin level gradually increases into the measurable range, although renin remains suppressed in some patients.15 Spironolactone or eplerenone may be added for blood pressure control if required. Measurement of ACTH is not useful for monitoring treatment, as normalisation may require glucocorticoid overreplacement. Similarly, normalisation of DOC and corticosterone levels usually reflects excess glucocorticoid dose.
Second, a sex steroid appropriate for the patient’s phenotypic sex is started at the expected time of puberty. Nearly all patients, regardless of genotypic sex, are raised as girls (with rare exceptions in XY patients with partial deficiencies and ambiguous genitalia). Replacement consists of sequential oestrogen followed by medroxyprogesterone, or the oral contraceptive pill. Low-dose testosterone may be added for sexual hair development.3
Lessons from practice
Unlike more common forms of congenital adrenal hyperplasia (CAH), CAH due to 17-α-hydroxylase deficiency may present in adulthood with hypertension and sexual infantilism.
The clinical presentation results from blockade of cortisol and sex steroid synthesis in the adrenals and gonads with resultant mineralocorticoid (ie, deoxycortisone) overproduction.
Treatment consists of physiological glucocorticoid replacement to normalise blood pressure and the potassium level, and replacement of sex steroids.
This is a rare but underdiagnosed disorder and should be considered in patients with primary failure of sexual development and hypertension.
The case we report highlights several salient clinical aspects of 17-α-hydroxylase deficiency. This rare form of CAH may be missed in childhood, as the classical features of CAH (hypotension, salt wasting and other features of cortisol insufficiency) are not present, and ambiguous external genitalia only occur with partial insufficiency in XY individuals. Hypertension and hypokalaemia are not routinely assessed in children, so delay of puberty may be the first clinical indication. It is important to monitor blood pressure when evaluating patients with delayed puberty. In our patient, it was a chance injury resulting in hospitalisation that finally led to the diagnosis, despite her previous presentation with typical clinical features. If untreated, 17-α-hydroxylase deficiency may lead to serious complications of life-threatening hypokalaemia, hypertension and osteoporosis. Medical practitioners should therefore be mindful of this rare but characteristic presentation of CAH.
2 Results of biochemical investigations on serum
ACTH = adrenocorticotrophic hormone. FSH = follicle-stimulating hormone. LH = luteinising hormone. DHEA = dehydroepiandrosterone. |
|||||||||||||||
3 Steroid synthesis in 17-α-hydroxylase deficiency
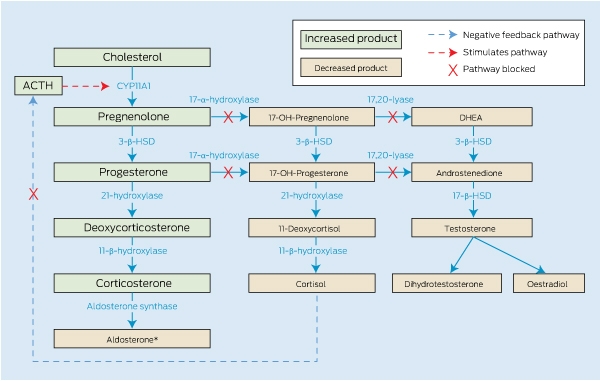
ACTH = adrenocorticotrophic hormone. CYP11A1 = side chain cleavage enzyme. DHEA = dehydroepiandrosterone. HSD = hydroxysteroid dehydrogenase. OH = hydroxy. * Aldosterone is usually suppressed, owing to the salt-retaining and volume expansion effects of the high concentrations of deoxycortisone, with consequent transcriptional downregulation of aldosterone synthase.
Competing interests
No relevant disclosures.
References
- Cameron FJ, Montalto J, Yong AB, Warne GL. Steroid 17alpha-hydroxylase deficiency: first Australian case report. J Paediatr Child Health 1997; 33: 259-261. 0_i1140660
- Takeda Y, Yoneda T, Demura M, et al. Genetic analysis of the cytochrome P-450c17alpha (CYP17) and aldosterone synthase (CYP11B2) in Japanese patients with 17alpha-hydroxylase deficiency. Clin Endocrinol (Oxf) 2001; 54: 751-758. 0_i1140662
- Kater CE. The 17-alpha and 11-beta hydroxylase deficiency forms of congenital adrenal hyperplasia. Presentation at ENDO 2011 (The Endocrine Society’s 93rd Annual Meeting); 2011 Jun 4-7; Boston, USA. 0_i1140664
- Human Gene Mutation Database [internet database]. Institute of Medical Genetics, Cardiff University. http://www.hgmd.cf.ac.uk/ac/index.php (accessed Sep 2012).
- Imai T, Yanase T, Waterman MR, et al. Canadian Mennonites and individuals residing in the Friesland region of The Netherlands share the same molecular basis of 17 alpha-hydroxylase deficiency. Hum Genet 1992; 89: 95-96. 0_i1140669
- Miura K, Yasuda K, Yanase T, et al. Mutation of cytochrome P-45017 alpha gene (CYP17) in a Japanese patient previously reported as having glucocorticoid-responsive hyperaldosteronism: with a review of Japanese patients with mutations of CYP17. J Clin Endocrinol Metab 1996; 81: 3797-3801. 0_i1140671
- Costa-Santos M, Kater CE, Auchus RJ; Brazilian Congenital Adrenal Hyperplasia Multicenter Study Group. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab 2004; 89: 49-60. 0_i1140673
- Jones KL, Freidenberg GR, Buchta R, Derenoncourt A. Male pseudohermaphroditism resulting from 17 alpha-monooxygenase (P-450C17) deficiency in two unrelated Guamanians. Am J Dis Child 1992; 146: 592-595. 0_i1140675
- Yang J, Cui B, Sun S, et al. Phenotype-genotype correlation in eight Chinese 17alpha-hydroxylase/17,20 lyase-deficiency patients with five novel mutations of CYP17A1 gene. J Clin Endocrinol Metab 2006; 91: 3619-3625. 0_i1140677
- Wei JQ, Wei JL, Li WC, et al. Genotyping of five Chinese patients with 17alpha-hydroxylase deficiency diagnosed through high-performance liquid chromatography serum adrenal profile: identification of two novel CYP17 mutations. J Clin Endocrinol Metab 2006; 91: 3647-3653. 0_i1140679
- Bao X, Ding H, Xu Y, et al. Prevalence of common mutations in the CYP17A1 gene in Chinese Han population. Clin Chim Acta 2011; 412: 1240-1243. 0_i1140681
- Wang W, Fu JF, Gong FQ, et al. Rare hypertension as a result of 17alpha-hydroxylase deficiency. J Pediatr Endocrinol Metab 2011; 24: 333-337. 0_i1140683
- Wang YP, Li J, Li JX, et al. Three novel CYP17A1 gene mutations (A82D, R125X, and C442R) found in combined 17-hydroxylase/17,20-lyase deficiency. Metabolism 2011; 60: 1386-1391. 0_i1140685
- New MI. Male pseudohermaphroditism due to 17 alpha-hydroxylase deficiency. J Clin Invest 1970; 49: 1930-1941. 0_i1140687
- Auchus RJ. The genetics, pathophysiology, and management of human deficiencies of P450c17. Endocrinol Metab Clin North Am 2001; 30: 101-119, vii. 0_i1140691