





Volume 197 - Issue 7
Severe drug-induced hypersensitivity syndrome with a shared HLA-B allele
Authors: Damian J Harding, Kavitha Subramaniam, Gerry MacQuillan, Joshua Davis and David Nolan
Med J Aust 2012; 197 (7): 411-413. || doi: 10.5694/mja12.10477
Published online: 1 October 2012
Published online: 1 October 2012
We describe the first reported cases of phenytoin-related drug-induced hypersensitivity syndrome in unrelated Indigenous Australians with the same HLA-B*56:02 allele. These patients shared common clinical and pathological features, and all three were found to have the otherwise rare HLA-B allele, which might represent a novel genetic susceptibility factor for this syndrome.Clinical recordsPatient 1A 32-year-old Indigenous man from a remote Western Australian community presented with a fever, ...
We describe the first reported cases of phenytoin-related drug-induced hypersensitivity syndrome in unrelated Indigenous Australians with the same HLA-B*56:02 allele. These patients shared common clinical and pathological features, and all three were found to have the otherwise rare HLA-B allele, which might represent a novel genetic susceptibility factor for this syndrome.
Clinical records
Patient 1
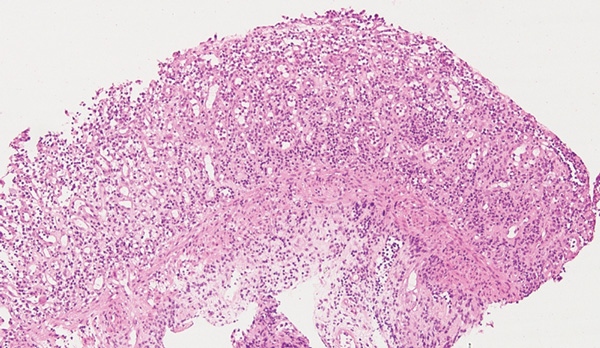
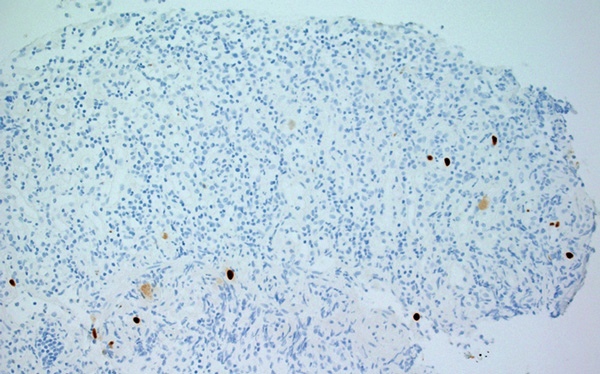
A 32-year-old Indigenous man from a remote Western Australian community presented with a fever, diffuse papular rash and oral ulceration, and a productive cough of 2 weeks’ duration. He was admitted to the intensive care unit (ICU) for respiratory failure and hypotension. Phenytoin 300 mg daily had been prescribed 3 weeks earlier for alcohol-associated seizures. He remained febrile with persistent respiratory failure, and after 10 days he developed profuse diarrhoea and transient hepatitis. Abdominal imaging results were normal and screening tests for bloodborne infections and autoimmune disease were negative. Colonoscopy showed severely inflamed, oedematous mucosa and confluent ulceration, with histological evidence of widespread mucosal loss and granulation tissue overlying inflamed superficial submucosa with prominent cytomegalovirus (CMV) inclusions (Box 1 and Box 2).
It was discovered that he had attended a different hospital 2 years earlier with a desquamating rash, oral ulceration, peripheral eosinophilia, lymphadenopathy and prolonged fever after starting phenytoin therapy. Delayed-onset features had included diarrhoea (with colonic CMV inclusions) and hepatitis. There was serological and polymerase chain reaction (PCR) evidence of human herpesvirus 6 (HHV-6) reactivation. A liver biopsy had shown features of drug hypersensitivity. After withdrawal of phenytoin treatment and starting treatment with prednisolone and valganciclovir, he had been discharged 47 days after admission, and had been advised to avoid phenytoin.
When this previous hospitalisation history came to our attention, we discontinued phenytoin treatment and started corticosteroid and ganciclovir treatment. There was no evidence of HHV-6 reactivation. CMV DNA was initially detected in colonic biopsies (on day 8), and subsequently in whole blood and stool samples (in week 5). The patient’s profuse diarrhoea persisted despite ganciclovir, opiate and octreotide treatment, and after 8 weeks it was complicated by recurrent bleeding requiring alternate-day blood transfusions. A further colonoscopy to investigate the bleeding showed regenerative, hypervascular colonic mucosa with reduced ulceration. After 11 weeks in the intensive care unit, the patient deteriorated with refractory hypotension and worsening respiratory failure, and died 2 days later.
Patient 2
A 33-year-old Indigenous man from Perth, Western Australia, presented with a generalised desquamating rash without mucosal involvement, and fever and abdominal pain of 4 weeks’ duration, 3 months after commencing phenytoin 300 mg daily for alcohol-related seizures. Investigations revealed hepatitis and eosinophilia, and gastroscopy showed gastric ulceration with prominent CMV viral inclusions. CMV and HHV-6 DNA were also detected in whole blood. Phenytoin treatment was ceased and corticosteroid and ganciclovir treatment was started, followed by oral valganciclovir until the CMV viraemia resolved. The patient recovered fully.
Patient 3
A 43-year-old Indigenous woman from a remote Northern Territory community presented with a cerebral abscess requiring surgical drainage and intravenous phenytoin prophylaxis 300 mg daily. She had an initially uneventful recovery until 2 weeks postoperatively, when she became progressively hypoxaemic, with bilateral pulmonary infiltrates. She developed a desquamating rash, hepatitis, eosinophilia and diarrhoea. Phenytoin treatment was ceased and she was admitted to the ICU. She developed progressive multiple organ failure and died 10 days later, despite receiving maximal supportive therapy.
Discussion
For all three patients, human leukocyte antigen (HLA) typing revealed HLA-B*56:02 as a common allele (including one patient with an HLA-B*56:02-homozygous allele). Although this allele is widely distributed globally, it is generally rare.1 Among Indigenous Australians, localised areas of high prevalence (more than 10%) have been found in central Australia, although previous studies of Indigenous populations in WA, and in Arnhem Land in the Northern Territory, suggest a population prevalence of less than 2%.1 Within the Australian Bone Marrow Donor Registry (which has recently been enriched by involvement of Indigenous Australian donors), HLA-B*56:02 is present in 0.6% (173/29 067) of donors who have had DNA sequencing performed at HLA-B.
The occurrence of this HLA-B allele in three unrelated cases therefore appears exceptionally unlikely to have occurred by chance alone, suggesting that it may be appropriate to add it to an existing list of HLA-B alleles marking genetic susceptibility to drug hypersensitivity syndromes. This list includes HLA-B*57:01 (hypersensitivity to abacavir and flucloxacillin), B*58:01 (allopurinol), B*15:02 (carbamazepine) and B*35:05 (nevirapine).2
These three cases are in keeping with diagnostic criteria for drug-induced hypersensitivity syndrome (DIHS) (formerly known as drug reaction with eosinophilia and systemic symptoms [DRESS] syndrome). Criteria include:
delayed-onset rash 2–6 weeks after initial drug exposure
haematological abnormalities (eosinophilia and/or atypical lymphocytosis)
systemic features with prolonged fever and/or lymphadenopathy as well as organ involvement (most frequently hepatitis, but also gastrointestinal, lung and kidney involvement).
Expanded diagnostic criteria acknowledge the diagnostic value of prolonged clinical symptoms 2 weeks after drug discontinuation. The prolonged clinical symptoms may reflect the role of herpesvirus reactivation in the perpetuation of hypersensitivity responses.3 Anticonvulsants are frequently implicated in DIHS, and in the case of carbamazepine-associated DIHS, there is evidence of HLA-associated susceptibility (in this case, with HLA-A*31:01).4
Patients may develop two or three features of the syndrome at one time, with symptom resolution in one organ often followed by development of symptoms in another organ system.5 DIHS and Stevens–Johnson syndrome share some features, including their delayed onset and prolonged course.6 However, mucocutaneous signs are more common in Stevens–Johnson syndrome, especially in the mouth and eyes, but facial oedema and small pustules are more typical of DIHS.7 In contrast, herpesvirus reactivation, hepatitis and other visceral involvement are more strongly associated with DIHS.
One unusual aspect of DIHS is the relationship between immune-mediated drug reactions and the reactivation of herpesviruses infection. This is particularly so for HHV-6 as well as CMV and Epstein–Barr virus (EBV), which may explain why organ involvement and systemic inflammation may be delayed in onset and prolonged after discontinuation of the offending drug. Real-time PCR analyses of subjects with DIHS have shown that various herpesviruses can sequentially reactivate during the syndrome, often initially HHV-6 or EBV, then HHV-7 and later CMV.8 Such a cascade of sequential reactivation is also seen in graft-versus-host disease.9 It seems likely in these cases that HLA-B*56:02 is involved in binding and presenting phenytoin (or a metabolite of phenytoin) to T-cells, although there are several possible mechanisms for HLA-restricted immune responses to drugs.10 In the context of DIHS, there is additional evidence that antiviral T-cells may also be affected by this process, so that delayed tissue injury may be mediated by pro-inflammatory antiviral immune responses rather than by the viruses themselves.5 The immunological basis for these reactions — and the temporal relationships between virus-specific and drug-specific immune responses — have not been completely elucidated. However, it is notable that B-cell depletion and hypogammaglobulinaemia are also found early in the clinical course of DIHS. One possible explanation for these B-cell abnormalities, in a syndrome that is otherwise thought to involve drug-specific T-cell responses, is that regulatory T-cell populations are preferentially involved in the early immune response.11
Mortality associated with DIHS is 10%–20%. Diagnostic awareness is therefore critical — including awareness of the potential for both delayed-onset and prolonged manifestations that may otherwise be viewed as atypical for a drug hypersensitivity syndrome. Previous experience with HLA-associated drug hypersensitivity reactions has demonstrated the contribution of genetic testing strategies towards improved vigilance and diagnostic accuracy. We believe that the novel finding of three patients with the same HLA-B*56:02 allele warrants further investigation in an expanded case series.
Competing interests
No relevant disclosures.
Acknowledgements
We thank Hooi Ee for help in reviewing and preparing this article.
References
- Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res 2011; 39: D913-D919. 0_i1140566
- Bharadwaj M, Illing P, Theodossis A, et al. Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annu Rev Pharmacol Toxicol 2012; 52: 401-431. 0_i1140568
- Shiohara T, Iijima M, Ikezawa Z, Hashimoto K. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations. Br J Dermatol 2007; 156: 1083-1084. 0_i1140570
- McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med 2011; 364: 1134-1143. 0_i1140572
- Kano Y, Ishida T, Hirahara K, Shiohara T. Visceral involvements and long-term sequelae in drug-induced hypersensitivity syndrome. Med Clin North Am 2010; 94: 743-759. 0_i1140574
- Teraki Y, Shibuya M, Izaki S. Stevens-Johnson syndrome and toxic epidermal necrolysis due to anticonvulsants share certain clinical and laboratory features with drug-induced hypersensitivity syndrome, despite differences in cutaneous presentations. Clin Exp Dermatol 2010; 35: 723-728. 0_i1140576
- Jeung YJ, Lee JY, Oh MJ, et al. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res 2010; 2: 123-126. 0_i1140578
- Shiohara T, Inaoka M, Kano Y. Drug-induced hypersensitivity syndrome (DIHS): a reaction induced by a complex interplay among herpesviruses and antiviral and antidrug immune responses. Allergol Int 2006; 55: 1-8. 0_i1140580
- Seishima M, Yamanaka S, Fujisawa T, et al. Reactivation of human herpesvirus (HHV) family members other than HHV-6 in drug-induced hypersensitivity syndrome. Br J Dermatol 2006; 155: 344-349. 0_i1140582
- Adam J, Pichler WJ, Yerly D. Delayed drug hypersensitivity: models of T-cell stimulation. Br J Clin Pharmacol 2011; 71: 701-707. 0_i1140584
- Takahashi R, Kano Y, Yamazaki Y, et al. Defective regulatory T cells in patients with severe drug eruptions: timing of the dysfunction is associated with the pathological phenotype and outcome. J Immunol 2009; 182: 8071-8079. 0_i1140588