





Volume 196 - Issue 7
Upper limb tremor
Authors: Thomas E Kimber and Philip D Thompson
Med J Aust 2012; 196 (7): 447-451. || doi: 10.5694/mja11.11565
Published online: 16 April 2012
Published online: 16 April 2012
Tremor is a common clinical problem in middle-aged and older patients, and Parkinson disease (PD) is one of the commonest causes. Careful history-taking and physical examination is usually sufficient for diagnosis of PD; extensive ...
Abstract
Tremor is a common clinical problem in middle-aged and older patients, and Parkinson disease (PD) is one of the commonest causes.
Careful history-taking and physical examination is usually sufficient for diagnosis of PD; extensive investigation is generally not required.
Treatment of PD should be individualised, taking into account the patient’s age, lifestyle, severity of motor symptoms, level of disability, comorbidities, expectations of treatment and PD subtype (eg, akinetic rigid or tremor dominant).
In PD, optimal medical therapy often involves a combination of dopaminergic medications, aiming for doses that provide adequate symptom relief without adverse effects such as dyskinesias and impulse-control disorders.
Continuous dopaminergic stimulation and deep brain stimulation should be considered for patients with PD whose motor symptoms cannot be adequately controlled with oral medication, especially those aged less than 70 years.
Robert’s story
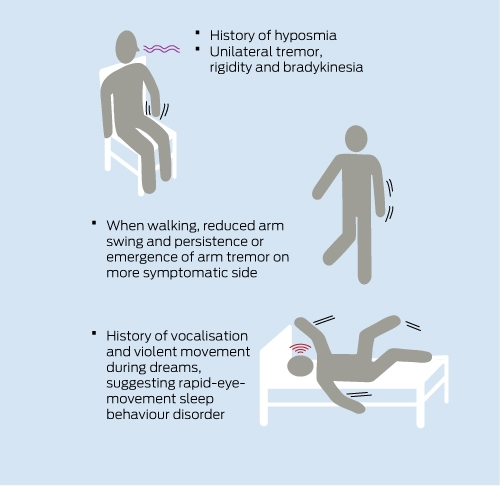
Robert is a 66-year-old man who presented to his general practitioner with a 6-month history of tremor in his left hand. He had noticed the tremor most while at rest and it was not particularly evident when he used the hand. He had, however, noticed that his hand was becoming less dextrous and he had been having trouble doing up buttons. The tremor was more evident while he was nervous or under pressure. He had also noticed some difficulty getting in and out of his car and turning over in bed. His wife commented that, over the previous 2 years, his posture had become more stooped.
On specific questioning, Robert reported that his sense of smell had been reduced for several years. His wife reported that he had been having unsettled sleep with vivid dreams, during which he would call out and thrash about. He had unwittingly hit her during some of these dreams.
On examination, Robert had a rather immobile face, with little spontaneous blinking. When he walked, there was noticeable asymmetry in his arm swing and tremor became evident in his non-swinging left arm. When he turned, he tended to stop and take several steps, with his shoulders and hips staying in the same plane during the turn. When he sat down, the tremor appeared in his left hand after a few seconds. He had a mild increase in muscle tone in his left upper limb that was consistent throughout the range of movement (“lead-pipe rigidity”) with superimposed regular variations (“cogwheel rigidity”). When he tapped his left fingers repetitively, the initial movements were relatively normal, but the movements then broke down into small amplitude movements that appeared clumsy and effortful. His eye movements were normal. He had normal strength, his tendon reflexes were symmetrical and normal, and he had flexor plantar responses. Results of a sensory examination were normal. His blood pressure was 170/90 mmHg lying and 150/80 mmHg standing.
Approach to the problem
Tremor is a common symptom in middle-aged and older patients. The differential diagnoses include primary neurological disorders, the commonest of which are essential tremor (ET), Parkinson disease (PD) and tremor induced by other medical conditions or medications (Box 1). In Robert’s case, a number of additional features indicate a likely diagnosis of PD (Box 2).
Characteristics of the movement disorder
Tremor
Several features of Robert’s condition are typical of parkinsonian tremor. This type of tremor is characteristically asymmetric and most evident at rest but may also persist (albeit usually at lesser amplitude) when the affected limb is elevated against gravity. Parkinsonian tremor typically diminishes or disappears during limb movement, such as a finger-to-nose test, or on changing posture; it then reappears after a short interval (re-emergent tremor). It is thus known as a rest, postural and re-emergent tremor. Parkinsonian tremor is characteristically evident in the affected arm during walking. An asymmetric rest tremor and good response to levodopa are the most specific diagnostic features of pathologically proven PD, although about 30% of patients with PD do not have tremor at presentation. Patients without tremor usually present with symptoms of bradykinesia.
By contrast, the tremor of ET affects both upper limbs more or less symmetrically and is a postural and action (kinetic) tremor — present when the limb is elevated against gravity and during movement, and absent or diminishing at rest. While the tremor of ET can be asymmetric, it is not unilateral, as is typically the case in early PD. Tremor of the head and voice are common in ET but not in PD; in PD, lip, chin or tongue tremor may be seen.
The distinction between PD and ET is not always straightforward. Patients with the tremor-dominant subtype of PD, which has a coarse resting tremor, are often misdiagnosed as having ET. In tremor-dominant PD, the clinical course is benign; there is little motor disability, rigidity or bradykinesia, and the tremor is relatively unresponsive to levodopa (depriving the clinician of one of the key features that aids accurate diagnosis). Diagnosis of PD is aided by clinical features additional to those described above, such as the presence of facial hypomimia or asymmetric arm swing when walking.
Another cause of upper limb tremor, which is less common than PD and ET, is adult-onset dystonic tremor. As in PD, dystonic tremor may be unilateral or asymmetric, and may occur at rest, during certain postures and during action. Helpful pointers to a diagnosis of dystonic tremor are position specificity or task specificity and other dystonic features such as abnormal posturing of the affected limb. Also, patients with dystonic tremor lack other features of PD, such as decremental bradykinesia, hyposmia and rapid-eye-movement sleep behaviour disorder (RBD).1,2
Other motor symptoms
Clinicians should enquire about symptoms of bradykinesia affecting the limbs, such as difficulty with repetitive or rotatory movements (eg, brushing teeth, washing hair) and with fine, coordinated movements (eg, handwriting, doing up buttons). It is also useful to ask about symptoms suggesting disordered axial movements, such as difficulty rising from a chair, getting out of a car or turning over in bed. These symptoms strongly suggest a parkinsonian disorder. In addition to tremor, the core clinical diagnostic features on examination include reduced spontaneous facial movement and blinking, giving a staring expression. Muscle tone is increased in the limbs and trunk. Bradykinesia is evident as a decrement in the amplitude of repetitive finger-tapping movements. A hypokinetic gait with short steps and reduced arm swing on the affected side may also be seen. Resting tremor may reappear in the affected upper limb during walking.
Certain red flags in the patient history and examination (Box 3) should raise doubt about the diagnosis of PD and suggest an alternative diagnosis such as multiple system atrophy or progressive supranuclear palsy (Box 4). The presence of such red flags should prompt referral to a neurologist for further assessment and investigation.
Non-motor symptoms
Non-motor symptoms are important for diagnosing a patient with tremor. It is common for non-motor symptoms — including constipation, sexual dysfunction, RBD, hyposmia, depression and anxiety — to predate the motor symptoms of PD. They may also constitute an important cause of disability in their own right.
RBD is characterised by the loss of normal muscle atonia during rapid-eye-movement sleep, causing the patient to “act out” dreams, which can lead to movements that injure the patient and their bed partner. At least 40% of patients with idiopathic RBD develop a neurodegenerative disorder at a mean interval of 11–12 years after onset of RBD.4,5 This is most commonly PD, multiple system atrophy or dementia with Lewy bodies (synucleinopathies).
Other questions to ask a patient with tremor
History-taking should also cover whether the patient has noticed a response of the tremor to alcohol. ET, especially when it is relatively mild, commonly lessens after alcohol intake, whereas this response is not seen in PD. Dystonic tremor may also be alcohol-responsive.1
Patients with tremor should also be asked whether they have a family history of tremor, which is present in at least 60% of cases of ET. However, a family history of tremor does not definitively distinguish ET from PD, as sporadic ET is common and PD may be familial.
Role of imaging
The diagnosis of PD is clinical. It is based on the presence of typical symptoms and signs, response to therapy and exclusion of other causes. In patients with typical clinical features and no red flags, cerebral imaging is not required. In patients with red flags that are atypical of PD, magnetic resonance imaging (MRI) of the brain should be used to look for radiological evidence of conditions such as multiple system atrophy, progressive supranuclear palsy and vascular parkinsonism.
Management
When to start treatment
None of the currently available medications for PD have been unequivocally shown to delay progression of the disease. For this reason, treatment for PD has traditionally been deferred until symptoms affect the patient’s quality of life, although this is currently being challenged by arguments in favour of earlier symptomatic treatment.
Clinical, imaging and neuropathological data suggest that the rate of progression of PD is faster in early disease than in late disease.6,7 Early dopaminergic treatment of PD is postulated to reduce the basal ganglia dysfunction that is associated with worsening motor symptoms.8 Some studies have suggested that early treatment with levodopa, dopamine agonists (eg, ropinirole, pramipexole) and the monoamine oxidase inhibitor rasagiline may have beneficial disease-modifying effects.9-12
There is accumulating evidence for the benefits of exercise and physical therapy in PD. In two recent studies, patients with PD who participated in a physical exercise program for 3–6 months exhibited benefits in executive cognitive function compared with controls.13,14 In addition, physical therapy may improve axial motor function, including balance, posture and gait, by an exercise-induced increase in endogenous dopamine release.15 Accordingly, patients with PD should be encouraged to engage in regular physical exercise.
Choice of initial symptomatic treatment
Initial symptomatic treatment should be individualised, taking into account the patient’s age, lifestyle, severity of motor symptoms, level of disability, comorbidities and expectations of treatment. In addition, the subtype and clinical expression of PD (eg, young onset, tremor dominant, akinetic rigid, or postural instability and gait disorder in older patients) may influence the initial approach to therapy. Two or three consultations may be needed to discuss all these points before making a decision about initial symptomatic treatment.
Levodopa with a dopa decarboxylase inhibitor (levodopa plus carbidopa or levodopa plus benserazide) is the most potent and effective drug therapy for the treatment of motor symptoms of PD (grade A evidence), and virtually all patients will require levodopa eventually. Nevertheless, initial therapy for PD has been a topic of vigorous debate in recent years. This has been prompted by concerns that most patients treated with levodopa will develop fluctuations in their motor response to levodopa after several years. The patient may notice a decline in motor benefit several hours after a dose of levodopa has been taken, which is known as the “end-of-dose” or “wearing-off” phenomenon. Around the same time, choreiform involuntary movements commonly emerge 1–2 hours after levodopa intake (peak-dose dyskinesias). Motor fluctuations and dyskinesias are believed to result from the declining capacity of the striatum to store dopamine in the dwindling population of pre-synaptic nigrostriatal neurones and the short plasma half-life of levodopa providing pulsatile (non-physiological) stimulation of striatal dopamine receptors. Younger patients (less than 60 years at diagnosis) are at greater risk of developing severe motor fluctuations,16 which may relate to a longer disease duration with greater depletion of nigrostriatal neurones and the tendency of younger patients to use large doses of medication. Concern has also been expressed that levodopa is toxic and that it loses efficacy over time, although there is no evidence to support either view.17 Finally, early treatment with dopamine agonists (as monotherapy or combined with levodopa) is associated with a lower incidence of motor fluctuations and dyskinesias in the short and medium term compared with levodopa monotherapy (grade A evidence). Long-term motor function, however, does not appear to be influenced significantly by initial treatment.18
Owing to the lower short- and medium-term risk of motor fluctuations, a dopamine agonist is preferred as initial treatment for patients who are younger than 70 years, with the later addition of levodopa as demanded by symptoms (usually within 2 years). In older patients, levodopa is often the better choice for initial treatment, because of the greater risk of adverse effects from dopamine agonists in older patients (eg, postural hypotension, delirium and hallucinations) and the greater improvement in motor function from levodopa. Levodopa should be introduced slowly and used at the lowest effective dose. Because motor improvement after starting levodopa therapy may evolve over several weeks, further assessment and any dose increases should occur after a minimum of about 3 months. Frequent changes to timing of medication and dose should be avoided and extra doses should be discouraged. Motor fluctuations and dyskinesias are more common when levodopa doses exceed 500–600 mg/day10 and it is noteworthy that the published incidence of motor fluctuations is derived from studies conducted in an era when higher doses of levodopa were commonly used. Accordingly, it is desirable to keep the dose of levodopa less than 600 mg/day for as long as possible. In practice, this means using a combination of a dopamine agonist and levodopa, and keeping the doses of both as low as practicable. It is possible that the incidence of severe motor fluctuations will be lower in the future, as lower levodopa dose regimens become more commonly used.
Complications of drug therapy
Adverse effects are an important consideration in the selection of treatment for early PD.
Postural hypotension, delirium and drowsiness
All dopaminergic medications can cause dose-dependent nausea, postural hypotension, delirium and drowsiness. These are more common with dopamine agonists than with levodopa and limit the dose that can be used.
Behavioural disturbances
Recently, attention has been focused on the behavioural disturbances that occur in up to 15% of patients with PD who are treated with high doses of dopaminergic medications.19,20 The most common are impulse-control disorders, which include pathological gambling, hypersexuality, compulsive buying and binge eating. The risk of these is increased in patients treated with dopamine agonists, either alone or in combination with levodopa.
Punding
Punding is a striking complication of dopaminergic therapy. It refers to the performance of repetitive and purposeless motor behaviour, such as collecting and rearranging objects and dismantling and reassembling equipment. Over-indulgence in hobbies — including internet use for excessive periods and at the expense of sleep, meals and other daily activities — also constitutes a form of punding. The incidence of punding in patients with PD is unclear, but it is thought to be less common than impulse-control disorders.19 Patients who begin dopaminergic therapy (especially dopamine agonist therapy) should be warned about the risk of impulsive and compulsive behaviour, and both they and their families should be asked to report any evidence of such behaviour.
Dopamine dysregulation syndrome
Another complication of dopaminergic therapy is dopamine dysregulation syndrome (DDS), which refers to the habitual use of dopaminergic medications at doses in excess of those prescribed and necessary for the treatment of motor symptoms. It appears to be less common than impulse-control disorders, occurring in about 4% of patients treated for PD.19 In patients with DDS, medication use seems to be dictated more by the mood elevation caused by medication and a desire to avoid the dysphoric effects of declining medication levels in “off” periods (ie, periods when PD symptoms are not adequately controlled) rather than by motor function.
Fibrotic reactions
Where possible, non-ergot dopamine agonists (eg, pramipexole, rotigotine) should be prescribed in preference to ergot agents (eg, bromocriptine, pergolide, cabergoline), because of the small risk of fibrotic reactions (eg, pleuropulmonary, retroperitoneal and cardiac valve fibrosis) associated with the latter.
Treatment of motor fluctuations and dyskinesias
In patients being treated with levodopa, wearing-off symptoms are usually managed by combining levodopa with additional dopaminergic medications, such as a catechol-O-methyltransferase inhibitor (eg, entacapone), dopamine agonist or a monoamine oxidase B inhibitor (eg, selegiline) (grade A evidence for all three). Slow-release levodopa preparations taken at bedtime may be useful for the treatment of wearing-off symptoms that occur overnight and on waking, but these have proven disappointing for daytime use because of variable absorption. Dividing the daily levodopa dose into smaller, more frequent aliquots and/or use of dispersible levodopa formulations may hasten absorption and assist with the relief of wearing-off symptoms. Apomorphine, an injectable dopamine agonist, can be given intermittently by subcutaneous injection for the relief of disabling off periods.
When the motor response to medication is suboptimal, patients can be advised to avoid taking medication with protein-containing meals, as amino acids compete with levodopa for transport across the blood–brain barrier.
Additional treatment options for patients with severe motor fluctuations and dyskinesias
For most patients with PD, a well tailored oral medication regimen provides adequate control of motor symptoms. The aim is to provide a regular, stable and continuous level of dopaminergic stimulation with as few changes as possible, avoiding booster doses. However, if motor fluctuations and dyskinesias become severe, or if oral medications are poorly tolerated, there are a number of other options, including continuous dopaminergic stimulation and deep brain stimulation.
Continuous dopaminergic stimulation
Options for continuous dopaminergic stimulation are apomorphine infusion and levodopa–carbidopa enteral gel infusion. Apomorphine infusion produces a substantial reduction in daily off time and a reduction in severity of dyskinesias (grade C evidence).21 Reduction in dyskinesias is particularly marked in patients for whom levodopa therapy is reduced or ceased.21 In one study, 70% of patients with PD who were commenced on apomorphine infusion were able to have other dopaminergic medications withdrawn and be managed with apomorphine monotherapy long term.21 However, another study reported a much lower rate (3%) of achieving apomorphine monotherapy and a discontinuation rate of 40% (due, in many cases, to adverse effects such as skin reactions, sedation and confusion).22
Infusion of levodopa–carbidopa intestinal gel directly into the jejunum produces more stable plasma levodopa levels than intermittent oral dosing.23 Intestinal levodopa infusion also leads to a significant reduction in off time, with either stable or reduced levels of dyskinesia (grade B evidence).24 Levodopa–carbidopa intestinal gel infusion is commenced in the inpatient setting, with a nasoduodenal tube, and the infusion rate is titrated according to therapeutic need. When the optimal motor response is achieved, a percutaneous gastrostomy tube with an inner jejunal tube is inserted for ongoing infusion. Patients receiving apomorphine and intestinal levodopa–carbidopa infusion need to be managed in a specialist PD clinic by an experienced multidisciplinary team, including a specialist PD nurse.
Deep brain stimulation
Deep brain stimulation of either the subthalamic nucleus or globus pallidus interna has an established role in the management of PD. Patients who have an excellent motor benefit from levodopa, but who have developed motor fluctuations and dyskinesias from long-term use of levodopa, are good candidates for deep brain stimulation, as are those with tremor that is refractory to medication. Deep brain stimulation produces a significant reduction in off time and substantially
reduces
tremor and dyskinesia (grade B evidence).25The benefits of deep brain stimulation in reducing tremor, off time, motor fluctuation and dyskinesia persist at up to 10 years of follow-up.26,27 However, there is some decline in objective measures of motor function with time. This decline appears to be due primarily to progression in domains of motor function that are not responsive to deep brain stimulation, such as freezing of gait in the on-medication state, postural instability and falls.26-28
Careful patient selection is critical to the success of deep brain stimulation. Patients with significant mood disorders or cognitive dysfunction do poorly. Severe postural instability, falls and freezing of gait in the on-medication state are not typically alleviated following deep brain stimulation and may in fact be made worse.28 Improvement in quality of life following deep brain stimulation is significantly greater in patients who are younger than 65 years compared with older patients, despite a similar motor benefit.29 This seems to be due to the greater incidence in older patients of cognitive dysfunction, postural instability and gait dysfunction.29 These problems are not reduced following deep brain stimulation and are major contributors to poor quality of life.
Fact or fiction?
Fact: It is true that virtually all patients with Parkinson disease will require levodopa eventually.
Fiction: It is not true that levodopa is toxic and loses effectiveness over time in Parkinson disease.
Robert was diagnosed with PD at his first visit to a neurologist. Discussion focused on whether to commence medication immediately or wait until more disabling problems occurred. Robert felt that his symptoms were not sufficiently severe to commence regular medication immediately. He was encouraged to keep physically active and adopt a regular exercise program. He was also encouraged to make contact with his local PD association, for information and support. For reputable and evidence-based information about PD, the WE MOVE website (http://www.wemove.org/par), affiliated with the international Movement Disorder Society, was recommended.
Six months later, Robert felt that his condition had deteriorated and wanted to commence treatment. Pramipexole extended release was prescribed by his neurologist, starting with 0.375 mg daily and increasing to 0.75 mg daily after 1 month. At a subsequent review, he reported a significant reduction in the tremor. The only adverse effect that he reported was a possible mild increase in drowsiness. He did not report any symptoms of impulse-control dysfunction.
One year after commencing pramipexole (and 18 months after after his first visit), Robert reported that his tremor was becoming worse. He had also noticed worsening manual dexterity with his left hand, slowing of his walking and greater difficulty rising from a chair and turning over in bed. Levodopa–carbidopa (100 mg levodopa plus 25 mg carbidopa, three times daily) was added to the pramipexole regimen, with a beneficial effect on these symptoms.
One year later (two and a half years after his first visit), Robert reported that he had noticed that the benefit from the levodopa–carbidopa tablets tended to wear off well before the next dose was due. His therapy was therefore changed from levodopa–carbidopa to levodopa–carbidopa–entacapone (100 mg levodopa plus 25 mg carbidopa plus 200 mg entacapone, three times daily), which resulted in a significant improvement in his condition. His pramipexole therapy was continued. He is being followed up by his neurologist every 6 months.
1 Differential diagnoses of tremor, and situations in which these occur
Parkinsonian tremor (rest, postural, re-emergent)
Essential tremor (postural, action)
Drug-induced tremor, which can be caused by valproate, antidepressants, antipsychotics, antiemetics, lithium, corticosteroids, cyclosporine, adrenergic agents, theophylline and caffeine
Dystonic tremor (postural, action, rest, task-specific)
Cerebellar (postural, action, intention)
Holmes (midbrain or rubral) tremor (rest, postural, action, intention)
Fragile X-associated tremor ataxia syndrome (rest, postural, action, intention)
Drug withdrawal
Thyrotoxicosis
Hypoglycaemia
Metabolic encephalopathies
Psychogenic tremor
4 Akinetic rigid syndromes to consider in a patient with parkinsonism and red flag features that are atypical of Parkinson disease
Multiple system atrophy
Progressive supranuclear palsy
Corticobasal degeneration
Dementia with Lewy bodies
Frontal white matter ischaemia (Binswanger disease)
Drug-induced parkinsonian tremor
Toxicity (eg, manganese, carbon monoxide, MPTP [1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine])
Prion disease
Competing interests
Thomas Kimber serves on Medical Advisory Boards for Hospira, Abbott and Biogen Idec and has received funding for travel from Novartis. Philip Thompson has served on the Medical Services Advisory Committee and on Medical Advisory Boards for Lundbeck, Boehringer Ingelheim, Novartis, Ipsen and Allergan; he has received funding for travel and an honorarium for educational lectures at a meeting sponsored by UCB Pharma; and he is Past President of the Movement Disorder Society.
References
- Schwingenschuh P, Schneider S, Silveira-Moriyama L, et al. Adult-onset dystonic tremor as differential diagnosis for Parkinson’s disease: detailed clinical description of 20 patients emphasizing non-motor symptoms including olfactory function. Parkinsonism Relat Disord 2007; 13 Suppl 2: S70-S71. 0_i1140700
- Schneider SA, Edwards MJ, Mir P, et al. Patients with adult-onset dystonic tremor resembling Parkinsonian tremor have scans without evidence of dopaminergic deficit (SWEDDs). Mov Disord 2007; 22: 2210-2215. 0_i1140702
- Hayes MW, Fung VS, Kimber TE, O’Sullivan JD. Current concepts in the management of Parkinson disease. Med J Aust 2010; 192: 144-149. 0_i1140705
- Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder. Neurology 1996; 46: 388-392. 0_i1140707
- Schenck CH, Bundlie SR, Mahowald MW. REM sleep behaviour disorder (RBD) delayed emergence of parkinsonism and/or dementia in 65% of older men initially diagnosed with idiopathic RBD, and an analysis of the maximum and minimum tonic and/or phasic electromyographic abnormalities found during REM sleep. Sleep 2003; 26 (Suppl): A316. 0_i1140709
- Schrag A, Dodel, Spottke A, et al. Rate of clinical progression in Parkinson’s disease. A prospective study. Mov Disord 2007; 22: 938-945. 0_i1140711
- Hilker R, Schweitzer K, Coburger S, et al. Nonlinear progression of PD as determined by serial positron emission tomographic imaging of striatal flurodopa F 18 activity. Arch Neurol 2005; 62: 378-382. 0_i1140713
- Schapira AH, Obeso J. Timing of treatment initiation in Parkinson’s disease: a need for reappraisal? Ann Neurol 2006; 59: 559-562. 0_i1140715
- Whone AL, Watts RL, Stoessl AJ, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol 2003; 54: 93-101. 0_i1140717
- Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med 2004; 351: 2498-2508. 0_i1140719
- Parkinson Study Group. Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA 2002; 287: 1653-1661. 0_pgfId-1154271
- Olanow CW, Rascol O, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361: 1268-1278. 0_i1140722
- Tanaka K, Quadros AC Jr, Santos RF, et al. Benefits of physical exercise on executive functions in older people with Parkinson’s disease. Brain Cogn 2009; 69: 435-441. 0_i1140724
- Cruise KE, Bucks RS, Loftus AM, et al. Exercise and Parkinson’s: benefits for cognition and quality of life. Acta Neurol Scand 2011; 123: 13-19. 0_i1140726
- Kwakkel G, de Goede CJT, van Wegen EEH. Impact of physical therapy for Parkinson’s disease: a critical review of the literature. Parkinsonism Relat Disord 2007; 13 Suppl 3: S478-S487. 0_i1140728
- Kempster PA, Williams DR, Selikhova M, et al. Patterns of levodopa use in Parkinson’s disease: a clinico-pathological study. Brain 2007; 130: 2123-2128. 0_i1140730
- Parkkinen L, O’Sullivan SS, Kuoppamaki M, et al. Does levodopa accelerate the pathologic process in Parkinson’s disease brain? Neurology 2011; 77: 1420-1426. 0_i1140732
- Katzenschlager R, Head J, Schrag A, et al. Fourteen-year final report of the randomized PDRG-UK trial comparing three initial treatments in PD. Neurology 2008; 71: 474-480. 0_i1140734
- Evans AH, Strafella AP, Weintraub D, Stacy M. Impulsive and compulsive behaviors in Parkinson’s disease. Mov Disord 2009; 24: 1561-1570. 0_i1140736
- Voon V, Hassan K, Zurowski M, et al. Prevalence of repetitive and reward-seeking behaviors in Parkinson disease. Neurology 2006; 67: 1254-1257. 0_i1140738
- Manson AJ, Turner K, Lees AJ. Apomorphine monotherapy in the treatment of refractory motor complications in Parkinson’s disease: longterm follow-up study of 64 patients. Mov Disord 2002; 17: 1235-1241. 0_i1140740
- Garcia Ruiz PJ, Ignacio AS, Pensado BA, et al. Efficacy of long-term continuous subcutaneous apomorphine infusion in advanced Parkinson’s disease with motor fluctuations: a multicentre study. Mov Disord 2008; 23: 1130-1136. 0_i1140742
- Nyholm D, Askmark H, Gomes-Trolin C, et al. Optimizing levodopa pharmacokinetics: intestinal infusion versus oral sustained-release tablets. Clin Neuropharmacol 2003; 26: 156-163. 0_i1140744
- Nyholm D, Nilsson Remahl AIM, Dizdar N, et al. Duodenal levodopa infusion monotherapy vs oral polypharmacy in advanced Parkinson disease. Neurology 2005; 64: 216-223. 0_i1140746
- Deuschl G, Schade-Brittinger C, Krack P, et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355: 896-908. 0_i1140748
- Moro E, Lozano AM, Pollak P, et al. Long-term results of a mulicenter study on subthalamic and pallidal stimulation in Parkinson’s disease. Mov Disord 2010; 25: 578-586. 0_i1140750
- Castrioto A, Lozano AM, Poon Y, et al. Ten-year outcome of subthalamic stimulation in Parkinson disease: a blinded evaluation. Arch Neurol 2011; 68: 1550-1556. 0_i1140752
- Rodriguez-Oroz MC, Obeso JA, lang AE, et al. Bilateral deep brain stimulation in Parkinson’s disease: a multicentre study with 4 years follow-up. Brain 2005; 128: 2240-2249. 0_i1140754
- Derost P, Ouchchane L, Morand D, et al. Is DBS-STN appropriate to treat severe Parkinson disease in an elderly population? Neurology 2007; 68: 1345-1355. 0_i1140756
Provenance: Commissioned; externally peer reviewed.