





Volume 192 - Issue 3
Current concepts in the management of Parkinson disease
Authors: Michael W Hayes, Victor S Fung, Thomas E Kimber and John D O’Sullivan
Med J Aust 2010; 192 (3): 144-149. || doi: 10.5694/j.1326-5377.2010.tb03453.x
Published online: 1 February 2010
Published online: 1 February 2010
Abstract
Parkinson disease (PD) is a multisystem neurodegenerative disorder that affects about 1% of the population over the age of 55 years and has mean age of onset of about 60 years.
The Braak hypothesis proposes that the earliest pathological evidence of PD is found in the enteric nervous system, medulla and olfactory bulb, and only subsequently progresses (over years) to the substantia nigra and cortex.
Non-motor symptoms, such as constipation, hyposmia and sleep disorders, may precede typical motor features of PD by several years.
No treatment has been convincingly shown to slow PD progression (ie, a neuroprotective drug remains elusive).
Symptomatic benefit from dopaminergic therapy is usually maintained throughout the course of the disease.
The decision as to whether to commence treatment with either levodopa or a dopamine agonist needs to be individually tailored, but long-term outcomes appear to be equivalent.
Advanced PD is complicated by the loss of non-dopaminergic neurones, resulting in symptoms that are largely unresponsive to dopaminergic therapy.
Treatment with apomorphine, Duodopa or deep-brain stimulation surgery may be beneficial for selected patients with advanced PD.
Non-motor symptoms, such as mood disorders, cognitive impairment, autonomic dysfunction and sleep disorders, are responsible for significant morbidity. Management often requires a multidisciplinary approach.
Parkinson disease (PD) is a heterogeneous multisystem neurodegenerative disorder with a selective vulnerability of dopaminergic neurones.1 It is a common condition, affecting about 1% of the population aged over 55 years. An estimated 60 000 Australians have PD, and about 10% of cases are diagnosed in people under the age of 50 years. The total economic cost of PD in Australia was estimated to be $6.8 billion in 2005.2
Recent advances in molecular biology have increased our understanding of pathogenesis. The discovery, in 1997, of an alpha-synuclein gene mutation in familial PD3 provided a “proof of principle” of a genetic contribution to PD, as it turned out that the encoded protein was a major component of the Lewy body, a pathological hallmark of PD.4 Although abnormal cellular protein aggregation and misfolding are common precursors to neuronal death, the function of the six identified familial PD genes indicates that this process may result from a wide range of cellular disorders.5 The cause of the much commoner sporadic or idiopathic PD is even less clear, but there is evidence that some monogenic variants or polymorphisms may represent susceptibility genes, conferring increased risk of PD. Many environmental risk factors for PD have been proposed, but epidemiological evidence suggests that the effects are relatively small.6
Detailed postmortem studies support the hypothesis of Braak and colleagues that the earliest pathological evidence of PD, as defined by the presence of Lewy bodies, starts in the enteric (gut) nervous system, medulla and olfactory bulb and spreads transneuronally to the midbrain (substantia nigra) and then the cortex.7 This may explain why non-motor symptoms of PD, such as constipation, hyposmia and rapid-eye-movement sleep disorder, often precede the typical motor symptoms by several years and why cognitive impairment is nearly always found in people with longstanding PD.8 Advancing PD is further complicated by the loss of non-dopaminergic neurones, contributing to disturbances of gait, posture, autonomic function, speech, cognitive function and sleep that may become unresponsive to dopamine.1 These developments underscore the difficulty of providing effective pharmacotherapy for patients with advanced PD and imply that dopamine replacement alone is ultimately inadequate.
Clinical features
Dopamine loss in the substantia nigra, which serves to modify motor control, results in the recognisable core signs of asymmetrical bradykinesia and hypokinesia (slowness and reduced amplitude of movement), muscle rigidity (stiffness) and rest tremor. Rest tremor, prominent asymmetry and a good response to levodopa (LD) are the features that most accurately predict PD pathology.9 The tremor-dominant form of PD tends to run a more benign course than typical PD. Early falls or autonomic symptoms and a questionable response to LD should raise doubts about the diagnosis (Box 1).10 Drug-induced parkinsonism due to commonly prescribed dopamine-blocking medications, such as antipsychotics (eg, haloperidol, risperidone) and antiemetics (eg, metoclopramide, prochlorperazine), should be excluded. Functional imaging of the dopaminergic system using cerebral single photon emission computed tomography or positron emission tomography can be useful in diagnosis of early or atypical PD (Box 2), but is unavailable in Australia. Positron emission tomography studies examining the rate of decline in dopamine-producing cells suggest that humans have already lost 50%–70% of their nigral neurones before they develop motor symptoms,11 and it has been estimated that the duration of this “presymptomatic” phase is about 5 years (Box 3). Early diagnosis will become a critical issue if effective neuroprotective drugs become available.
Treatment principles in early Parkinson disease
The early management of PD is based on the following principles:
The main aim of treatment is to preserve quality of life, which can be adversely affected by motor or non-motor symptoms;
There are many effective symptomatic treatments for PD;
No treatment has been convincingly shown to slow or hasten disease progression;13 and
A symptomatic response to dopaminergic therapy is usually maintained throughout the disease.
Note that the levels of evidence referred to here are determined by the American Academy of Neurology and the European Federation of Neurological Societies (A = established as effective, B = probably effective, C = possibly effective) (Box 4).10,14,15,17,18
Treatment of motor symptoms in early Parkinson disease
Although PD is a progressive disorder, deterioration is typically very slow, with considerable individual variability. Satisfaction with the explanation of the diagnosis has been shown to have a favourable impact on the quality of life of people with PD in the long term.19 The time to commence drug treatment for motor symptoms is when they are causing physical or psychological disability. It is a misconception that PD treatment is only effective for a limited time and should be deferred for as long as possible to reserve that benefit. Most movement disorder specialists also believe that physical therapy is important, although there is limited evidence to support this.20
Levodopa versus a dopamine agonist as first-line therapy
LD combined with a dopa-decarboxylase inhibitor (LD/DDI) remains the most potent drug therapy for reversing motor impairment (Level A). Despite its short half-life of about 90 minutes, dosing with LD/DDI three times daily usually provides a stable response without symptom fluctuations in early PD. At this stage, LD has a long-duration (pharmacodynamic) effect in the brain that is independent of plasma levels. A higher maintenance dose of LD (eg, 200 mg three times daily compared with an initial dose of 100 mg three times daily) provides slightly greater benefit for reducing motor symptoms, but at the cost of earlier wearing-off symptoms and dyskinesias (see below).
Initiating treatment with a dopamine agonist (DA) (a synthetic dopamine receptor agonist with a longer plasma half-life than LD/DDI) reduces the risk of dyskinesias and wearing-off fluctuations in the first few years of therapy (Level A evidence for cabergoline and pramipexole; Level B for pergolide and bromocriptine). However, DAs are less potent than LD for ameliorating motor symptoms, and most people require the addition of LD/DDI within 2 years. At that stage, there is still a rationale for ongoing combination therapy, as DAs may serve as an LD dose-sparing agent as well as offering pharmacokinetic advantages. Rotigotine, a non-ergoline (non-ergot-derived) DA delivered by transdermal 24-hour patch, is available in Australia but is not yet funded by the Pharmaceutical Benefits Scheme (PBS). Its efficacy is similar to that of oral DAs.
All dopaminergic medications can cause nausea, gastrointestinal symptoms, hypotension, drowsiness, cognitive symptoms and impulse control disorders (see below), but these are more common with DAs than LD/DDIs. The ergoline DAs can also cause clinically significant fibrosis (cardiac valvular, pleuropulmonary and retroperitoneal), a problem avoided by the newer, non-ergoline DAs (Box 4). There is no distinctive difference between the DAs in clinical efficacy. In general, there is a trend towards initiating treatment with a DA in people with earlier-onset PD and using LD/DDI as first choice in people with later-onset PD (Box 5). Two studies comparing long-term outcomes (> 10 years) in patients initially treated with either DA or LD/DDI failed to identify any ultimate advantage of one strategy over the other.21,22
Disease-modifying therapy
No treatment has been proven unequivocally to slow disease progression in PD. There has been renewed interest in the role of monoamine oxidase type B (MAO-B) inhibitors (eg, selegiline), which inhibit catabolism of dopamine. A recent study using a more potent MAO-B inhibitor, rasagiline (not available in Australia), in previously untreated PD patients suggested a small, sustained benefit in patients who began treatment earlier compared with those who started the drug 9 months later.23 Several earlier studies of MAO-B inhibitors, also using delayed-start protocols, had shown similar modest effects, but interpretation of these findings is controversial. The hypothesis that early treatment, regardless of which agent is used, may delay subsequent irreversible compensatory mechanisms in the basal ganglia is of current interest.24
Later treatment of Parkinson disease
Fluctuations and dyskinesias — pathogenesis
Motor fluctuations and dyskinesias are believed to be caused by progressive loss of striatal dopamine storage capacity and the pulsatile fashion in which LD is administered.25 Fluctuations in the motor response to LD usually emerge after several years of treatment.26 The patient may notice a decline in motor benefit several hours after the last dose of LD has been taken (“end-of-dose deterioration” or “wearing-off phenomenon”). Choreiform involuntary movements commonly emerge within 1–2 hours after LD intake (peak-dose dyskinesias). Later, dyskinesias may also occur at the onset and offset of motor benefit (diphasic dyskinesias).
Fluctuations and dyskinesias — management
Wearing-off symptoms are usually managed by combining LD with additional dopaminergic medication, such as a catechol-O-methyltransferase inhibitor (entacapone) (Level A), a DA (Level A), or an MAO-B inhibitor (selegiline) (Level C). Slow-release LD preparations (eg, Sinemet CR [Merck Sharp and Dohme, Sydney, NSW] and Madopar HBS [Roche, Sydney, NSW]) may be useful at bedtime to relieve nocturnal akinesia and early morning dystonia, but are hampered during the day by unpredictable gut absorption. Fractionating the daily LD dose into smaller, more frequent doses and administering liquid LD/DDI formulations (eg, Madopar Rapid [Roche, Sydney, NSW]) may hasten absorption and reduce “off” time (ie, periods of time when PD symptoms are not adequately controlled) (Level C). As amino acids compete with LD for transfer across the blood–brain barrier, separating the times of dietary protein and LD intake may improve drug effectiveness. Amantadine (100–300 mg daily) can help to suppress peak-dose dyskinesias (Level C).
For most patients with PD, motor fluctuations and dyskinesias are not disabling and can be adequately managed by manipulating the oral drug regimen. However, some patients with PD (particularly those with early-onset disease) develop severe motor fluctuations and dyskinesias, and alternative routes of medication delivery or surgical treatment may be considered. There is experimental and clinical evidence that motor fluctuations can be reduced with the use of continuous administration of dopaminergic medication.27 Apomorphine is an injectable DA that is administered either as intermittent subcutaneous bolus injections or by subcutaneous infusion. Apomorphine treatment produces a reduction in daily off time of about 50%,28,29 and long-term benefit can be maintained in some patients.28 However, side effects, including skin nodules, can be problematic.
More stable plasma levels of LD, and a corresponding reduction in motor fluctuations, can be achieved by delivering LD/DDI directly into the jejunum via percutaneous gastrostomy.30 A concentrated gel form of LD–carbidopa (Duodopa [Solvay Pharmaceuticals, Hannover, Germany]) has been developed for this purpose and successfully trialled in Europe.31 Duodopa has been approved by the Therapeutic Goods Administration in Australia, but is expensive and not PBS-funded. Patients receiving apomorphine and Duodopa need to be managed in specialist clinics by an experienced multidisciplinary team.
Neurosurgical treatments for PD have gained increasing importance in recent years. Lesioning (eg, pallidotomy and thalamotomy) has largely been superseded by deep brain stimulation (DBS) surgery. This technique involves subcutaneous implantation of a pulse generator or pacemaker(s) below the clavicle (usually bilaterally), with a connecting lead and electrodes that are stereotactically placed in the targeted brain site(s). In appropriately selected patients with advanced PD, DBS of the subthalamic nucleus or globus pallidus interna significantly reduces off-time motor symptoms, reduces dyskinesias and improves quality of life (Level B, although there is sufficient recent evidence for Level A).32-35 The beneficial effects of DBS on certain motor symptoms (such as tremor, bradykinesia and dyskinesias) appear to be long-lasting.32 However, other symptoms, including gait dysfunction and falls, do not necessarily improve after DBS and may even worsen.35 Appropriate patient selection for DBS is critical, and Australian consensus guidelines have been recently published.36
Non-motor symptoms
A variety of non-motor features can contribute significantly to morbidity and may be resistant to, and even aggravated by, standard dopaminergic therapy. The tendency for dopaminergic therapy to aggravate orthostatic hypotension and visual hallucinations in advancing PD creates a common management dilemma. Disturbances of sensation (including pain perception and olfaction), autonomic function, sleep, mood, behaviour and cognition occur frequently, and are difficult to manage.
Autonomic dysfunction
Orthostatic hypotension, constipation, and disturbance of bladder function, erectile function and sweating occur frequently in PD. Although constipation can be an early feature of PD, prominent dysautonomia early in the course of parkinsonism raises the possibility of multiple system atrophy (Box 1). Management involves drug treatment and non-pharmacological strategies, but these have not been subjected to large clinical trials, particularly in patients with PD (Box 6).
Mood disturbance
Depression, apathy and anxiety can occur in the early stages of PD, possibly related to lowered serotonin levels, and treatment with antidepressants or anxiolytics may be necessary before treatment of motor symptoms.37 An overlap between depressive and parkinsonian symptoms can delay recognition and treatment of both depression and PD. Depression has a major impact on quality of life in PD, possibly more so than motor disability.19 Anxiety can be particularly troublesome in patients with more advanced PD, with symptoms sometimes paralleling motor fluctuations. There is some evidence that treatment with a DA may reduce depression.14,15 Limited controlled trials have demonstrated that tricyclic antidepressants (including nortriptyline and amitriptyline [Level C]) but not selective serotonin reuptake inhibitors are effective for treating depression in patients with PD.17,38 Nevertheless, we have found selective serotonin reuptake inhibitors to be effective in clinical practice.
Sleep disturbance
Overnight motor and autonomic symptoms, including dystonia and frequent nocturia, are common in people with PD, but primary disorders of sleep may also occur. Rapid-eye-movement sleep behaviour disorder, characterised by violent limb and trunk movements and often associated with dream enactment, occurs in about 30% of patients with PD and has emerged as a marker of early PD.37 Restless legs syndrome occurs frequently in patients with PD and often responds to dopaminergic therapy. Excessive daytime somnolence is common, and may be disease- or treatment-related.
Neuropsychiatric symptoms
A range of neuropsychiatric symptoms can be triggered or aggravated by drug therapy in patients with PD. Psychotic symptoms, particularly visual hallucinations, are more common in those with cognitive decline or dementia.37 A reduction of anti-parkinsonian medication is often necessary. Clozapine is effective for reducing psychotic symptoms (Level B), but its use is limited by side effects14,15 and, like other antipsychotics, it is not reimbursed under the PBS for this indication. There is insufficient evidence to support the use of other atypical antipsychotics. Quetiapine is relatively safe and may help (Level C), but olanzapine and especially risperidone often aggravate parkinsonism, even at low doses.14,15
Impulse control disorders, including pathological gambling and hypersexual behaviour, have recently been recognised as an important side effect of dopaminergic medication in up to 15% of patients with PD.39 DAs (both ergoline and non-ergoline) are more commonly implicated than LD, and dose reduction or cessation usually improves this behaviour. Dopamine dysregulation syndrome, a less common condition, is related to medication overuse and involves more complex and challenging behavioural problems. Dopaminergic reward pathways in the limbic region have been implicated.39
Cognitive symptoms and dementia
The incidence of dementia increases with duration of PD. It is characterised by fluctuating cognition and visual hallucinations. Cognitive impairment affects up to 75% of people who have had PD for at least 15 years, although the main risk factor is advancing age.8 If cognitive symptoms emerge within 12 months of the motor symptoms, the condition is described as “dementia with Lewy bodies” rather than “PD dementia”, although pathologically the two conditions are similar. Clinical trials with cholinesterase inhibitors have demonstrated benefit in treatment of cognitive symptoms (Level A) and behavioural and psychotic symptoms (Level B) in patients with both dementia with Lewy bodies and PD dementia.14,15,17,40 However, they are not PBS-approved for this indication in Australia.
1 Features not typical of primary or idiopathic Parkinson disease (PD)10
Acute onset
Rapid progression
Early falls
Poor response to levodopa*
Symmetrical clinical signs
Early autonomic symptoms
Vertical gaze palsy
Early cognitive impairment
* Of the cardinal features of PD, tremor is normally the least responsive to levodopa.
2 Clinical hallmarks of some atypical parkinsonian syndromes
Progressive supranuclear palsy
Early falls
Axial rigidity
Supranuclear gaze palsy
Blepharospasm
Frontalis muscle overactivity (“startled” appearance)
Usually symmetrical limb signs
No response or poor response to levodopa
3 Loss of neurones with progression of Parkinson disease (PD)*
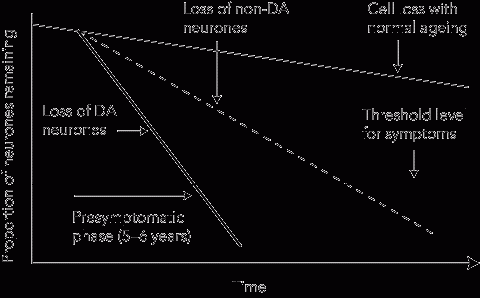 |
|
DA = dopaminergic. * This conceptual figure assumes that loss of DA and non-DA neurones begins at the same time. However, there is evidence of selective vulnerability of DA neurones in PD, and hence more rapid loss of these cells and a predominance of DA symptoms (eg, motor impairment) in early PD. Neuroprotective or restorative treatments aimed at DA neurone loss alone are unlikely to have much impact on non-DA symptoms (eg, cognitive impairment), which tend to predominate in advanced PD. Neuroprotective strategies targeting primary neurodegenerative mechanisms as well as vulnerable DA neurones may be required. (Figure adapted, with permission, from Lang.12) |
4 Pharmacological and non-pharmacological treatments for Parkinson disease (PD): regulatory status and evidence for use
5 Suggested treatment algorithm for Parkinson disease (PD)
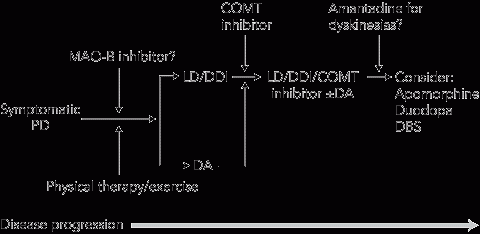 |
|
COMT = catechol-O-methyltransferase. DA = dopamine agonist. DBS = deep brain stimulation. DDI = dopa-decarboxylase inhibitor. LD = levodopa. |
6 Management of autonomic symptoms
Increase dietary salt, water, caffeine
Compression stockings
Head-up tilt in bed
Fludrocortisone 0.1–0.3 mg daily
Midodrine 2.5–10 mg daily
Domperidone 10–30 mg daily
Pyridostigmine 30–90 mg daily
Urinary frequency/nocturia/detrusor hyperreflexia
Oxybutynin 5–15 mg daily
Tolterodine 2–4 mg daily
Amitriptyline or nortriptyline 10–30 mg daily (beware of orthostatic hypotension and constipation)
Avoid anticholinergic drugs
Ensure adequate intake of fluid, fibre and fruit
Add laxatives (macrogol)
Erectile dysfunction
Competing interests
Michael Hayes is on advisory boards for Novartis and Solvay; Victor Fung and John O’Sullivan are on advisory boards for Novartis, Solvay, Hospira and Boehringer Ingelheim; and Thomas Kimber is on advisory boards for Solvay and Hospira.
References
- Lang AE, Obeso JA. Current challenges in Parkinson’s disease: restoring the nigro-striatal dopamine system will not suffice. Lancet Neurol 2004; 3: 309-316. 0_i1092405
- Living with Parkinson’s disease: challenges and positive steps for the future. Canberra: Access Economics for Parkinson’s Australia, 2007. http://www.parkinsons.org.au/media-advocacy/docs/pd-study.pdf (accessed Oct 2009).
- Polymeropoulos MH, Lavendan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997; 276: 2045-2047. 0_i1092409
- Spillantini MG, Schmidt MI, Lee VM, et al. Alpha synuclein in Lewy bodies. Nature 1997; 388: 839-840. 0_i1092411
- Gasser T. Update on the genetics of Parkinson’s disease. Mov Disord 2007; 22 Suppl 17: S343-S350. 0_i1092413
- Predborski S. Etiology and pathogenesis of Parkinson’s disease. In: Jankovic J, Tolosa E, editors. Parkinson’s disease and movement disorders. 5th ed. New York: Lippincott Williams & Wilkins, 2006: 77-92. 0_i1092415
- Braak H, Del Tredici K, Rüb U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24: 197-211. 0_i1092417
- Hely M, Reid W, Adena M, et al. The Sydney Multicenter Study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord 2008; 23: 837-844. 0_i1092419
- Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002; 125: 861-870. 0_i1092421
- Suchowersky O, Reich S, Perlmutter J, et al. Practice parameter: diagnosis and prognosis of new onset Parkinson disease (an evidence-based review): report of the Quality Standards Committee of the American Academy of Neurology. Neurology 2006; 66: 968-975. 0_i1092423
- Morrish PK, Sawle GV, Brooks DJ, et al. The rate of progression of Parkinson’s disease. A longitudinal [18F]DOPA PET study. Adv Neurol 1996; 69: 427-431. 0_i1092425
- Lang AE. The progression of Parkinson disease: a hypothesis. Neurology 2007; 68: 948-952. 0_i1092427
- Suchowersky O, Gronseth G, Perlmutter J, et al. Practice parameter: neuroprotective strategies and alternative therapies for Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006; 66: 976-982. 0_i1092429
- Horstink M, Tolosa E, Bonuccelli U, et al. Review of the therapeutic management of Parkinson’s disease. Report of a joint task force of the European Federation of Neurological Societies (EFNS) and the Movement Disorder Society — European Section (MDS-ES). Part I: early (uncomplicated) Parkinson’s disease. Eur J Neurol 2006; 13: 1170-1185. 0_i1092431
- Horstink M, Tolosa E, Bonuccelli U, et al. Review of the therapeutic management of Parkinson’s disease. Report of a joint task force of the European Federation of Neurological Societies (EFNS) and the Movement Disorder Society — European Section (MDS-ES). Part II: late (complicated) Parkinson’s disease. Eur J Neurol 2006; 13: 1186-1202. 0_i1092433
- Brainin M, Barnes M, Baron JC, et al. Guidance for the preparation of neurological management guidelines by EFNS scientific task forces — revised recommendations 2004. Eur J Neurol 2004; 11: 577-581. 0_i1092437
- Miyasaki JM, Shannon K, Voon V, et al. Practice parameter: evaluation and treatment of depression, psychosis, and dementia in Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006; 66: 996-1002. 0_i1092438
- Pahwa R, Factor S, Lyons K, et al. Practice parameter: treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006; 66: 983-995. 0_i1092440
- Global Parkinson’s Disease Survey Steering Committee. Factors impacting on quality of life in Parkinson’s disease: results from an international survey. Mov Disord 2002; 17: 60-67. 0_i1092442
- Keus S, Munneke M, Nijrake M, et al. Physical therapy in Parkinson’s disease: evolution and future challenges. Mov Disord 2009; 24: 1-14. 0_i1092444
- Katzenschlager R, Head J, Schrag A, et al. Fourteen-year final report of the randomized PDRG-UK trial comparing three initial treatments in PD. Neurology 2008; 71: 474-480. 0_i1092446
- Hely MA, Morris JG, Reid WG, et al. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa responsive problems dominate at 15 years. Mov Disord 2005; 20: 190-199. 0_i1092448
- Rascol O, Olanow CW. The effects of rasagiline in early Parkinson’s disease: the ADAGIO delayed start trial. Mov Disord 2009; 24 Suppl 1: S275. 0_i1092450
- Schapira AH, Obeso JA. Timing of treatment initiation in Parkinson’s disease: a need for reappraisal? Ann Neurol 2006; 59: 559-562. 0_i1092452
- Obeso JA, Rodriguez-Oroz M, Marin C, et al. The origin of motor fluctuations in Parkinson’s disease: importance of dopaminergic innervation and basal ganglia circuits. Neurology 2004; 62 Suppl 1: S17-S30. 0_i1092454
- Schrag A, Quinn N. Dyskinesias and motor fluctuations in Parkinson’s disease. A community based study. Brain 2000; 123: 2297-2305. 0_i1092456
- Stocchi F, Olanow CW. Continuous dopaminergic stimulation in early and advanced Parkinson’s disease. Neurology 2004; 62 Suppl 1: S56-S63. 0_i1092458
- Hughes A, Bishop S, Kleedorfer B, et al. Subcutaneous apomorphine in Parkinson’s disease: response to chronic administration for up to five years. Mov Disord 1993; 8: 165-170. 0_i1092460
- Pietz K, Hagell P, Odin P. Subcutaneous apomorphine in late stage Parkinson’s disease: a long term follow up. J Neurol Neurosurg Psychiatry 1998; 65: 709-716. 0_i1092462
- Nyholm D, Askmark H, Gomes-Trollin C, et al. Optimizing levodopa pharmacokinetics: intestinal infusion versus oral sustained-release tablets. Clin Neuropharmacol 2003; 26: 156-163. 0_i1092464
- Antonini A, Isaias IU, Canesi M, et al. Duodenal levodopa infusion for advanced Parkinson’s disease: 12-month treatment outcome. Mov Disord 2007; 22: 1145-1149. 0_i1092466
- Rodriguez-Oroz MC, Obeso JA, Lang AE, et al. Bilateral deep brain stimulation in Parkinson’s disease: a multicentre study with 4 years follow-up. Brain 2005; 128: 2240-2249. 0_i1092468
- Deuschl G, Schade-Brittinger C, Krack P, et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355: 896-908. 0_pgfId-1092470
- Rodrigues JP, Walters SE, Watson P, et al. Globus pallidus stimulation in advanced Parkinson’s disease. J Clin Neurosci 2007; 14: 208-215. 0_pgfId-1092471
- Weaver FM, Follett K, Stern M, et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson’s disease: a randomized controlled trial. JAMA 2009; 301: 63-73. 0_i1092472
- Silberstein P, Bittar RG, Bole R, et al. Deep brain stimulation for Parkinson’s disease: Australian referral guidelines. J Clin Neurosci 2009; 16: 1001-1008. 0_i1092474
- Chaudhuri KR, Healy DG, Schapira AH; National Institute for Clinical Excellence. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 2006; 5: 235-245. 0_i1092476
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson’s disease and depression. Neurology 2009; 72: 886-892. 0_i1092478
- O’Sullivan SS, Evans AH, Lees AJ. Dopamine dysregulation syndrome: an overview of its epidemiology, mechanisms and management. CNS Drugs 2009; 23: 157-170. 0_i1092480
- Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004; 351: 2509-2518. 0_i1092482