




Cystic fibrosis (CF) is the most common life-shortening, autosomal recessive disease affecting people of European ancestry.1 It results in multisystem disease, predominantly affecting the lungs and the gastrointestinal system.
Previously, it was reported that the incidence of CF in Australia was about 1 in 2800 live births2 and about 1 in 25 people were carriers of a genetic mutation responsible for CF.3
Overseas studies have shown that, in the 1960s, median survival time with CF was about 10 years but is now more than 35 years.4,5 Factors thought to have contributed to improved survival are newborn screening, management of patients in specialist CF centres, improvement in treatment of neonatal meconium ileus and pancreatic insufficiency, better antibiotics, and more effective approaches to nutrition and airway clearance.1
In 1998, Cystic Fibrosis Australia (CFA) supported the development of a data registry in Australia and New Zealand, based on the American Cystic Fibrosis Foundation Patient Registry.4 The purpose of the registry was to allow measurement of the demographics, morbidity and mortality of the CF population to increase awareness about CF, improve quality of patient care through comparisons of CF centres, and facilitate research. People with CF are identified through centres that also coordinate data submission. Recommended practice in Australia is for people with CF to be referred to a CF centre, which is usually a multidisciplinary specialist team attached to a teaching hospital.6
Data for this report were extracted from information submitted by all CF centres in 2009. Patients who reside in the Northern Territory and Australian Capital Territory have shared-care with neighbouring states, and their data are submitted by the CF centre that they attend. Demographic and diagnosis information, pulmonary function test results, anthropometric measurements, microbiological culture findings, hospitalisation rates, and information on treatments, complications, mortality and transplantation were collected. If multiple entries had been made for a patient, the measurement of best pulmonary function and the highest values for height and weight for the year were used. Standard deviation scores (z scores) were calculated using United States National Center for Health Statistics and Centers for Disease Control and Prevention normalised growth reference values for height, weight and body mass index (BMI). We only included pulmonary function test results for patients over 6 years of age and used two pulmonary function prediction equations that were appropriate for age.7,8 We recorded microbiological culture findings for all organisms isolated from respiratory cultures performed during the year.
We estimated incidence using 5-year birth cohorts for 2004–2008 and births data from the Australian Bureau of Statistics.9 We considered patients to be children up to 11 years of age, adolescents if they were 12–17 years and adults from age 18 years. We reported children’s nutritional status for the 2–5-years and 6–11-years age groups. The Student t test was used to compare lung function, z scores and BMI. P < 0.05 was considered to be statistically significant.
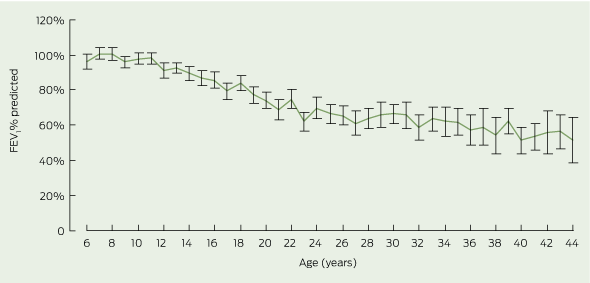
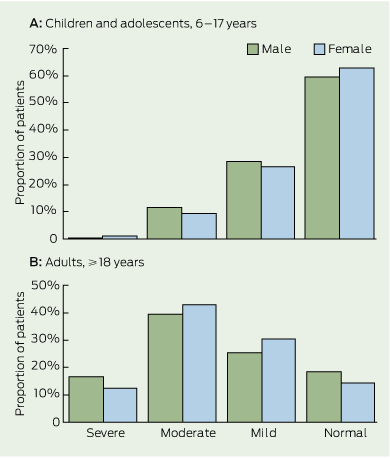
Percentage predicted forced expiratory volume in one second (FEV1 % predicted) decreased with increasing age (Box 1). The mean FEV1 % predicted for children and adolescents aged 6–17 years was 92.8% (SD, 18.2%) and for adults was 66.4% (SD, 22.2%). More than 50% of children and adolescents, but only 15% of adults, had an FEV1 % predicted in the normal range (Box 2). Notably, there was a wide range of lung function in all age groups. FEV1 % predicted did not differ by sex for children (boys, 92.9% [SD, 18.3%]; girls, 92.7% [SD, 18.2%]; P = 0.87) or for adults (men, 66.0% [SD, 23.3%]; women, 68.8% [SD, 20.8%]; P = 0.55).
Specimens for microbiological culture were mostly sputum across all age groups, but samples from bronchoscopy were available for 26% of children. Methicillin-sensitive Staphylococcus aureus infection was common among all age groups, and Haemophilus influenzae infection was also common among children (Box 3). The prevalence of Pseudomonas aeruginosa infection increased with increasing age. Burkholderia cepacia complex and methicillin-resistant S. aureus (MRSA) infection were relatively uncommon among all age groups (Box 3).
Overall, young children aged 2–5 years were found to be normally nourished, but with increasing age, z scores for both weight and height decreased (Box 4). Adolescent males were more poorly nourished than adolescent females, with lower z scores for weight (P = 0.05) and BMI (P = 0.02). Mean BMI for adults was 22.5 kg/m2 (SD, 3.3). BMI was higher in men than in women (23.1 kg/m2 [SD, 3.3] v 21.9 kg/m2 [SD, 3.2]; P < 0.001). Mild undernutrition (BMI, 18 to < 20 kg/m2) was seen in 13.3% of men and 20.0% of women. Severe undernutrition (BMI, < 18 kg/m2) was evident in 3.7% of men and 7.8% of women.
With increasing age of patients, there was an increase in the frequency of pulmonary complications such as haemoptysis, pneumothorax, and respiratory failure (Box 5). A similar pattern was seen with gastro-oesophageal reflux (both symptomatic and endoscopically proven), abnormal biochemical liver function tests, cirrhosis and pancreatitis. CF-related diabetes (intermittent) requiring insulin therapy was not seen in children. It was evident as a complication among adolescents and was most common in adults. Low bone density increased with age, yet the fracture rate for all age groups was low.
Attendances for outpatient review were relatively constant over all age groups. The proportion of patients hospitalised for all CF-related indications was similar in children aged 2–5 years compared with those aged 6–11 years, but higher for adolescents and adults (Box 6). Of all patients admitted, 82% (897/1091) were admitted for respiratory complications. The proportion of patients requiring two or more respiratory-related hospitalisations was higher among adolescents (115/502; 23%) and adults (167/748; 23%) compared with children (148/1034; 14%).
There are about 3200 people with CF in Australia, as we estimate that the 2986 patients identified are about 90% of the total number of people with CF in Australia. The incidence of CF in Australia now approaches one in 3000 births. Most new diagnoses of CF in 2009 were made through the Australia-wide newborn screening program. However, diagnosis is still delayed in some children born since implementation of the screening program, and four people were diagnosed as adults. Early diagnosis is expected to be associated with improved outcomes.10
While we have estimated that the ACFDR covered 90% of people with CF in 2009, some underrepresentation of patients was evident in the ACFDR report, and reporting has varied.11,12 Our estimate is based on opinions of CF centres and comparisons with other data. Comparisons between total deaths by age group in the ACFDR with any ICD-10 (International Classification of Diseases, 10th revision) code indicating CF from the Australian Institute of Health and Welfare (AIHW) National Mortality Database suggest some under-coverage in adult age groups, possibly due to loss to follow-up when private care is chosen, or after lung transplantation, for which care is exclusively at transplant centres. We plan to include analysis of supplementary data from transplant centres in future registry reports. Our confidence in the completeness of data for Australian children with CF is based on the system of referral to CF centres following diagnosis through newborn screening programs. With the enthusiasm and goodwill of many, complete population coverage including all adults and those who have undergone lung transplantation will be achieved within several years. The overall level of coverage achieved in Australia compares favourably with that of overseas data registries.13 Recent improvements have been online data entry and the generation of patient reports suitable for clinical care. Data registries need to meet a quality standard to be effective, and this requires significant resources.14 While the ACFDR is well established, annual data collection is an unfunded burden for CF centres and failure to collect data would threaten the registry’s contribution to further improving outcomes for people with CF through research and better care.
The development of data registries allows comparison of outcomes between CF centres within countries and internationally to identify practices that will enhance health. For example, the finding that children with CF from Victoria had better survival than UK patients15 was attributed to the model of coordinated specialised centre care in Victoria and led to the development of specialist CF care centres worldwide. The most recent study of life expectancy among patients with CF in Australia found that from 1979 to 2005 the mean age at death increased from 12.2 years to 27.9 years for males and from 14.8 to 25.3 years for females.16 The ACFDR has attempted only a preliminary survival analysis because of uncertainty about ascertainment of all deaths (unpublished data); however, they have begun a project comparing deaths reported to the ACFDR with deaths reported to the AIHW National Mortality Database to allow more accurate survival analyses. Centre-to-centre comparisons are now well established in the US, and have been available for Australian CF centres for the past 3 years.17 However, their validity is dependent on there being complete and high-quality data and standardised methods for comparison of health status.13 This requires resources that are not readily available for this activity in CF centres.14
Progressive respiratory disease is the major cause of morbidity and mortality among people with CF. Despite the absence of symptoms of CF, there is evidence of pulmonary disease early in life. Bronchoscopy specimens have shown evidence of airway infection and inflammation in young children with CF, and computed tomography scans have shown bronchiectasis in 22% of preschool children with CF.18,19 FEV1 is the single best predictor of mortality.20 Our data indicate that FEV1 is relatively normal between 6 and 11 years of age, with an increase in the rate of decline in FEV1 between 12 and 25 years. This is similar to the pattern of maximal decreases in FEV1 in adolescence shown by US data.21 Targeted interventions for children before they enter adolescence may be the key to establishing further improvements in morbidity and survival associated with CF.
This evidence for more severe respiratory disease in adolescence is associated with a doubling of the prevalence of P. aeruginosa infection among adolescents compared with children. Chronic P. aeruginosa infection is a risk factor for poorer outcome.22,23 Implementation of strategies to avoid chronic P. aeruginosa infection earlier in childhood may lead to improved lung function in adulthood. The rates of isolation of MRSA and B. cepacia complex are low but increase with age. Cross-infection between patients is a potential problem for CF centres, as these organisms are associated with poorer outcomes and can be a contraindication for transplantation, so implementation of infection control policies is standard practice.24
A consequence of improved life expectancy is that 50% of people with CF are now adults. Adulthood among patients with CF is associated with more severe lung disease, poorer nutritional status and more CF-related complications such as haemoptysis, pneumothorax, diabetes, metabolic bone disease and the consequences of male infertility.v1 Despite this burden of disease and associated treatment demands, we found that most adults with CF work or study and many establish relationships and families. As it is estimated that more than 90% of children with CF will reach adulthood, caring for adolescents requires a focus which allows for minimal interruption of schooling while allowing people with CF to achieve their potential.5 The CF care team needs to consider flexible access to care for the individual who is continuing to study, work and develop lifelong relationships.25 Helping adolescents and young adults to accept demanding treatment programs is a challenge for CF teams.
People with CF require lifelong medical support, and new adult-specific CF centres have emerged over the past 25 years. Recently published Australian guidelines for CF care6 recommend that patients be reviewed quarterly; but, according to the ACFDR, half the population received fewer reviews than this. Furthermore, hospitalisation in 2009 equated to about 3000 inpatient episodes (about 30 000 occupied bed-days), of which 80% were for treatment of respiratory complications. The national annual health system cost for CF care has been estimated to be around A$67 million.26 The distribution of costs included inpatient costs, 58%; pharmaceuticals, 29%; medical services, 10%; complications, 2%; and diagnostic tests, 1%. ACFDR data indicate that the burden of care increases as patients age, so CF health care planning is urgently required — particularly given the increase in numbers of adults with CF.11,12 The forthcoming national health reforms provide an opportunity for a coordinated approach to health service planning for CF.27
3 Findings from microbiological culture of sputum or bronchoscopy samples* from Australian patients with cystic fibrosis, 2009
* Number and percentage of patients with at least one culture positive for the organism. |
|||||||||||||||
Received 7 June 2011, accepted 17 August 2011
- Scott C Bell1
- Peter T P Bye2
- Peter J Cooper3
- A James Martin4
- Karen O McKay3
- Phillip J Robinson5
- Gerard F Ryan6
- Geoff C Sims7
- 1 Department of Thoracic Medicine, The Prince Charles Hospital, Brisbane, QLD.
- 2 Department of Respiratory Medicine, Royal Prince Alfred Hospital, Sydney, NSW.
- 3 Department of Respiratory Medicine, The Children’s Hospital at Westmead, Sydney, NSW.
- 4 Department of Respiratory Medicine, Women’s and Children’s Hospital, Adelaide, SA.
- 5 Department of Respiratory Medicine, Royal Children’s Hospital, Melbourne, VIC.
- 6 Department of Respiratory Medicine, Sir Charles Gairdner Hospital, Perth, WA.
- 7 Australian Cystic Fibrosis Data Registry, Cystic Fibrosis Australia, Sydney, NSW.
We acknowledge the support of each of the participating CF centres, including centre directors, CF care coordinators and data entry personnel; CFA for financial support, and their staff, in particular, Mr Terry Stewart, Chief Executive Officer; state CF organisations for additional financial support; Roche Pharmaceuticals and Solvay Pharmaceuticals for past and present financial support.
Geoff Sims has received consultancy fees for the management of the ACFDR. Scott Bell, James Martin, Phillip Robinson, Gerard Ryan and Geoff Sims have received support from CFA for travel and, when required, accommodation to attend ACFDR Medical Advisory Committee Meetings held in Sydney.
- 1. O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet 2009; 373: 1891-1904.
- 2. Massie RJ, Olsen M, Glazner J, et al. Newborn screening for cystic fibrosis in Victoria: 10 years’ experience (1989–1998). Med J Aust 2000; 172: 584-587.
- 3. Massie J, Forbes R, Dusart D, et al. Community-wide screening for cystic fibrosis carriers could replace newborn screening for the diagnosis of cystic fibrosis. J Paediatr Child Health 2007; 43: 721-723.
- 4. Cystic Fibrosis Foundation. Patient registry, 2008 annual data report. Bethesda: CFF, 2011.
- 5. Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 2007; 29: 522-526.
- 6. Fitzgerald DA, ed. Cystic fibrosis standards of care. Sydney: Cystic Fibrosis Australia, 2008.
- 7. Wang X, Dockery DW, Wypij D, et al. Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol 1993; 15: 75-88.
- 8. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general US population. Am J Respir Crit Care Med 1999; 159: 179-187.
- 9. Australian Bureau of Statistics. Births, Australia, 2009. Canberra: ABS, 2010. (ABS Cat. No. 3301.0.)
- 10. Southern KW, Merelle MM, Dankert-Roelse JE, Nagelkerke AD. Newborn screening for cystic fibrosis. Cochrane Database Syst Rev 2009; (1): CD001402.
- 11. Cystic Fibrosis Australia. Cystic fibrosis in Australia, 1998. Annual report from the Australian Cystic Fibrosis Data Registry. Sydney: CFA, 2000.
- 12. Cystic Fibrosis Australia. Cystic fibrosis in Australia, 2009. 12th annual report from the Australian Cystic Fibrosis Data Registry. Sydney: CFA, 2011.
- 13. McCormick J, Sims EJ, Green MW. Comparative analysis of Cystic Fibrosis Registry data from the UK with USA, France and Australasia. J Cyst Fibros 2005; 4: 115-122.
- 14. Evans SM, Bohensky M, Cameron PA, McNeil J. A survey of Australian clinical registries: can quality of care be measured? Intern Med J 2011; 41: 42-48.
- 15. Phelan P, Hey E. Cystic fibrosis mortality in England and Wales and in Victoria, Australia 1976–80. Arch Dis Child 1984; 59: 71-73.
- 16. Reid DW, Blizzard CL, Shugg DM, et al. Changes in cystic fibrosis mortality in Australia, 1979–2005. Med J Aust 2011; 195: 392-395. <MJA full text>
- 17. Sims G. International benchmarking from centre-level cystic fibrosis data. Pediatr Pulmonol 2010; 45 (S33): 410.
- 18. Ranganathan SC, Parsons F, Gangell C, et al. Evolution of pulmonary inflammation and nutritional status in infants and young children with cystic fibrosis. Thorax 2011; 66: 408-413.
- 19. Stick SM, Brennan S, Murray C, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr 2009; 155: 623-628.e1.
- 20. Kerem E, Reisman J, Corey M, et al. Prediction of mortality in patients with cystic fibrosis. N Engl J Med 1992; 326: 1187-1191.
- 21. Liou TG, Elkin EP, Pasta DJ, et al. Year-to-year changes in lung function in individuals with cystic fibrosis. J Cyst Fibros 2010; 9: 250-256.
- 22. Emerson J, Rosenfeld M, McNamara S, et al. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 2002; 34: 91-100.
- 23. Kerem E, Corey M, Gold R, Levison H. Pulmonary function and clinical course in patients with cystic fibrosis after pulmonary colonization with Pseudomonas aeruginosa. J Pediatr 1990; 116: 714-719.
- 24. Orens JB, Estenne M, Arcasoy S, et al. International guidelines for the selection of lung transplant candidates: 2006 update — a consensus report from the Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2006; 25: 745-755.
- 25. Tuchman LK, Schwartz LA, Sawicki GS, Britto MT. Cystic fibrosis and transition to adult medical care. Pediatrics 2010; 125: 566-573.
- 26. Van Gool K, Norman R, Delatycki MB, et al. Understanding the costs of care for cystic fibrosis: an analysis by age and severity. (Working Paper 2011/1.) Sydney: Centre for Health Economics Research and Evaluation, University of Technology Sydney, 2011.
- 27. Conway SP, Stableforth DE, Webb AK. The failing health care system for adult patients with cystic fibrosis. Thorax 1998; 53: 3-4.
Abstract
Objectives: To describe the demographics, clinical features and outcomes among people with cystic fibrosis (CF) in Australia and to estimate incidence of the disease.
Design and setting: Cross-sectional analysis using data from the Australian Cystic Fibrosis Data Registry for 2009.
Main outcome measures: Numbers of diagnoses, pulmonary and anthropometric measurements, microbiological culture results, rates of hospitalisation and transplantation, and numbers of medical complications and deaths.
Results: In 2009, data were submitted on 2986 people (48% female). Median age was 17.6 years and 49% of people were aged 18 years or over. Seventy-eight people were newly diagnosed. Fourteen people died and 14 people underwent lung transplantation in the year. Lung function and nutrition were relatively normal among children but deteriorated (more rapidly) among adolescents. With increasing age, progressive respiratory disease was apparent, and the frequency of CF-related complications and use of health care resources increased. In all age groups, there was a wide range in severity of lung disease and nutritional status.
Conclusions: CF remains a progressive respiratory disease and is associated with multisystem complications. The acceleration in disease severity in adolescence and early adulthood suggests that better treatment at these stages is required to further improve survival.