





Volume 195 - Issue 7
Changes in cystic fibrosis mortality in Australia, 1979–2005
Authors: David W Reid, C Leigh Blizzard, Dace M Shugg, Ceri Flowers, Catherine Cash and Hugh M Greville
Med J Aust 2011; 195 (7): 392-395. || doi: 10.5694/mja10.11229
Published online: 3 October 2011
Published online: 3 October 2011
Abstract
Objective: To assess mortality trends among people with cystic fibrosis (CF) in Australia.
Design and setting: We augmented Australian summary data for deaths from CF registered during 1979–2005 with information from Australian transplant centres on lung transplantation among CF patients for 1989–2005 to allow us to follow trends in all “mortality events” (death or lung transplantation).
Main outcome measure: Age at death or lung transplantation.
Results: Between 1979 and 2005, the mean age at death increased from 12.2 years to 27.9 years for males and from 14.8 years to 25.3 years for females. Overall, female deaths in childhood (0–14 years) occurred at an age-standardised rate of 0.40 per 100 000 (95% CI, 0.34–0.45) during 1979–2005, which exceeded the corresponding rate for males of 0.24 (95% CI, 0.20–0.28) per 100 000. Among 0–14-year-old boys, event rates declined markedly after 1989, but they declined later and more gradually for girls, with the result that the age-standardised rate for girls was 2.38 times that of boys during 1989–2005 (95% CI, 1.69–3.36).
Conclusions: The pattern of CF mortality in Australia has changed substantially. Mortality rates continue to be higher for girls than for boys, but death in childhood has become uncommon. Survival has increased since 1979, but females continue to have reduced length of life.
Survival among people with cystic fibrosis (CF) has increased worldwide over the past three decades. In countries where patients have access to specialised CF centres, the number of people with CF aged 18 years or over now equals or even exceeds that of the paediatric population. Before 1970, many individuals with CF died in infancy and, globally, the median age at death was only 8 years. However, with the introduction of multidisciplinary care teams and specialised CF centres, survival around the world began to improve. Newborn screening has allowed earlier diagnosis and intervention, and it is anticipated that this development will be reflected in further improvements in outcome. Current estimates are that, for patients with access to best CF centre care, more than 90% of the CF population born from 1990 onwards will reach the age of 40 years and the median survival of infants in the United Kingdom who currently have CF will exceed 50 years.1
Most patients with CF in Australia now have access to care in CF centres and, consistent with reports from other countries, are living to greater ages. However, there has been no formal assessment of survival and how it has changed over recent decades. In this study, we have analysed information on deaths or lung transplantation (“mortality events”) among the Australian population with CF during 1979–2005 to determine whether Australia has benefited from improvements in survival to the same extent as has been reported for the United States and Europe. We have also compared mortality event rates and age at death or lung transplantation between males and females to determine whether the sex imbalance that has historically favoured males, particularly during childhood and adolescence, is disappearing in Australia, as has been suggested elsewhere.2-8
Methods
Sources of data
Summary mortality data were sourced from the state and territories GRIM (General Record of Incidence of Mortality) book for cystic fibrosis (code E84, International Classification of Diseases, 10th revision), version 2, 2005, which was produced by the Australian Institute of Health and Welfare (AIHW). The GRIM provided data on deaths from CF each year for the period 1997–2005, stratified by 5-year age group and sex. Data for the years 1979–1996 were added to the GRIM by the AIHW at our request.
The lung transplant centres in Australia, located at the Alfred Hospital in Melbourne, St Vincent’s Hospital in Sydney, and the Prince Charles Hospital in Brisbane, provided de-identified data for 299 patients with CF who had received lung transplants during 1989–2005, including 177 who were still alive on 31 December 2005. Age at transplantation was considered a mortality event for the purposes of this study. Access to post-transplant age at death allowed us to cross-validate AIHW mortality data. Although lung transplantation prolongs life for patients with CF, the focus of this study is on CF mortality with respect to survival gains as a consequence of improved CF care; in this context, transplantation is a confounder. Age at transplantation is a reliable surrogate for age at death, as the median survival in patients with severe disease who are being considered for lung transplantation is about 1 year.9
The Australian Bureau of Statistics (ABS) supplied mid-year population estimates stratified by 5-year age group, sex and state or territory for each year of the period 1979–2005.
Cross-validation and collation
of data
We identified 25 individuals who had died following lung transplantation but were not recorded in the mortality dataset because CF had not been listed as a cause of death. We also identified 177 CF patients who had received a lung transplant before 31 December 2005 and had survived to 31 December 2005. This process of cross-validation enabled us to make adjustments to the summary data tabulated in the 2005 GRIM book for CF.
We also discovered that 25 people registered as having died of CF at the age of 60 years or over were incorrectly listed as CF deaths owing to coding errors. Therefore, no individuals over the age of 60 years were included in the mortality analysis.
Ethics approval
The study was approved by the Tasmania Health and Medical Human Research Ethics Committee and the human research ethics committees of the Alfred Hospital, Melbourne, St Vincent’s Hospital, Sydney, and the Prince Charles Hospital, Brisbane.
Statistical analysis
We calculated rates of mortality events (death or lung transplantation) by dividing annual or aggregated counts by annual or aggregated mid-year population estimates. We used the direct method to calculate age-standardised rates, applying age- and sex-specific rates to the Australian census population in 2001. This population is used as the standard in the 2005 GRIM book for CF. We also calculated female-to-male ratios of age-standardised rates (standardised rate ratios). Ninety-five per cent confidence limits for the rates, age-standardised rates and rate ratios were calculated using large-sample approximations. We used Poisson regression to estimate trends in rates, and linear regression to estimate trends in age-standardised rates.
To calculate mean age at time of death or lung transplantation from the summary mortality data, age was assigned as the mid-point of each 5-year age group. The data were transformed before analysis to remove positive skewness. We estimated trends in mean rates by linear regression.
Results
We identified 1072 people (511 males, 561 females) as having died with CF during 1979–2005 in Australia. Deaths occurred at an age-standardised rate of 0.25 per 100 000 population (95% CI, 0.24–0.27).
During 1989–2005, 299 (163 male, 136 female) CF patients received lung transplants and 177 (95 male, 82 female) of these survived to the end of 2005. Taken together, the two mortality events (death or lung transplantation) occurred at an age-standardised rate of 0.30 per 100 000 population (95% CI, 0.28–0.31).
Age-standardised rates of mortality and mortality events for the “pre-transplantation” period (1979–1988) and the “transplantation” period (1989–2005) are shown in Box 1. Overall, female mortality rates were a little higher than male rates. The female-to-male ratio of death rates was 1.13 (95% CI, 0.92–1.39) during 1979–1988, and 1.11 (95% CI, 0.96–1.29) during 1989–2005.
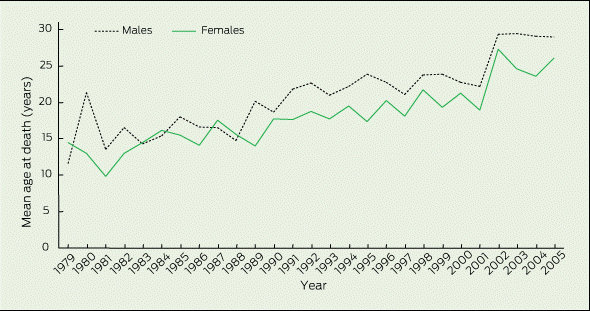
Trends in mean age at death (age at death in the absence of transplantation, or age at transplantation) during 1979–2005 are shown in Box 2. In most years, the mean age at death for males exceeded that for females. Overall, mean age at death increased from 13.3 years (95% CI, 12.2–14.4) in 1979 to 26.6 years (95% CI, 25.6–27.7) in 2005. This was an annual rate of increase of 187 days per year (95% CI, 160–213). Considered separately, the mean age at death for males increased from 12.2 years (95% CI, 13.5–16.0) in 1979 to 27.9 years (95% CI, 26.0–29.1) in 2005. For females, the increase was from 14.8 years (95% CI, 11.0–13.3) to 25.3 years (95% CI, 24.1–26.5). This equated to an overall average male-to-female difference in mean age at the mortality event of 941 (95% CI, 541–1341) days. If we consider this sex gap during 1989–2005, the average male survival advantage was even higher, at 1310 days (95% CI, 868–1752).
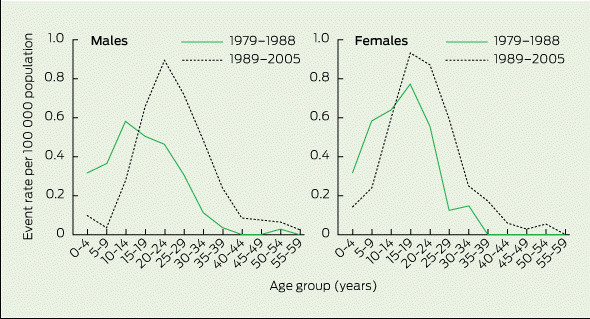
Age-specific rates for mortality events are shown in Box 3. For both sexes, a marked shift occurred in the later time period, with reduced mortality rates for children aged 0–9 years and boys aged 10–14 years, accompanied by higher mortality rates for older people. Notably, the mortality rate for girls aged 10–14 years remained unchanged.
From 1989 onwards, event rates decreased markedly among 0–14-year-old boys, but decreased much more gradually for girls over the entire period from 1979 to 2005. There were too few transplants in patients younger than 15 years of age (one boy, three girls) for the observed declines to be attributable to lung transplantation. There were corresponding increases in event rates over the same period for people aged 15–39 years (P < 0.01 for each sex).
Female-to-male ratios of age-standardised rates of mortality events by age group are provided in Box 4. The rate among girls aged 0–14 years was 1.6 times the rate for boys over the entire surveillance period (P < 0.05). However, the greater decline in mortality rate for boys (67%) compared with girls (36%) between 1989 and 2005 resulted in the mortality rate for girls being 2.4 times that for boys (P < 0.05). For older individuals, the male rate was higher than the female rate during 1989–2005, but this simply reflected the shift in mortality in males from childhood to adulthood during this period.
Discussion
Our analysis shows that mean age at death for the population of people with CF in Australia rose progressively during the 1979–2005 study period at a rate of increase of around 190 days per year. The increase in survival was predominantly related to a postponement of deaths that previously occurred during childhood years. This shift was most pronounced among children aged 0–14 years. However, even at the end of the study period, the mortality rate in girls aged 0–14 years remained more than twice that of boys. Thus, whereas males with CF who died during 1979–1988 lived on average around 460 days longer than females with CF, this survival gap had increased to around 1300 days during 1989–2005 (1200 days if lung transplantation is not taken into account).
Several reports from the US, UK and Europe have highlighted the increase in survival of individuals with CF over the past 30 years,1,10-13 but the Australian experience has not been formally documented. In our study among Australians with CF, mean age at death in 2005 was 26.6 years, which is slightly older than that reported for the same year in a German study (mean age at death 23.7 years).14 Cystic Fibrosis Data Registry reports in the US documented a median age at death of 25.3 years for 2005.15 In the UK, the median age at death in 2004 was 25.6 years.16 These reports suggest that Australian outcomes are comparable with other developed countries with high-quality health care systems. An international comparison of median age at death between 1975 and 2000 found that there was considerable variability in outcomes between countries.13 The study included data from Australia, but the analysis did not look at mortality rates in specific age cohorts, nor did it examine sex differences.
A factor that has been overlooked in previous studies of CF mortality is the confounding effect of lung transplantation. Our data suggest that if age at transplantation is not considered as age at death for statistical purposes, then CF survival may not be estimated accurately on a population basis — and this will be particularly true of countries that have a comprehensive transplant program. The improved survival of people with CF around the world has been attributed to the introduction of multidisciplinary care located in CF centres.
The reduction in mortality in our study was predominantly observed among children, and this experience is replicated in other countries.5,11,17 Whereas more than half of all CF deaths in Australia occurred in those aged younger than 15 years in 1979–1981, this age group accounted for only 5% of deaths in 2003–2005. Improved survival in childhood is linked to a doubling of the mortality rate for the 15–39-years age group. At the other end of the spectrum, survival past the age of 39 years was a rarity before 1989, but it has slowly become more common since 1988, particularly among males. This suggests that we are beginning to see the delayed effects of better care in childhood 25 years ago, building on the gains in childhood survival that were first noted in Australia in 1984.18 The net result has been an increase in the adult population with CF such that, according to the Australian national data registry in 2005,19 41% of the CF population in Australia are now adults aged 18 years and over, and this is likely to increase rapidly over the next decade.
Previous studies have described a sex difference in CF mortality, but most of these did not look at the mortality rate in specific age groups.2-5,20 One study from 1992 showed that females with CF aged younger than 20 years had a 60% increased risk of death compared with their male counterparts.2 A similar study reported that the overall risk of death across all ages was 30% higher in females.21 Nearly all studies of mortality in CF report a consistently higher average age at death for males, although more recent data from Germany suggest that this sex imbalance may be disappearing, at least in that country.14 Our study demonstrates that a sex difference persists in Australia, and that this became more marked in the late 1980s, particularly in terms of childhood deaths.
A striking finding of our study was how mortality trends for boys and girls aged 0–14 years changed over time, with a marked decline in event rates for boys after 1986 that was accompanied by a far more gradual decline in rates for girls. Our data cannot inform us of the cause of the earlier and more pronounced reduction in childhood mortality for boys. A potential explanation for a reduction in childhood mortality overall was the introduction of newborn screening in Australia. Newborn screening was first introduced in New South Wales in 1981. By 1990, it had become available in all states other than Western Australia.22,23 Early diagnosis has been shown to improve outcomes and reduce hospitalisations in children with CF, and there are suggestions that screening may reduce early mortality by up to 10%.24 The problem with attributing the reduction in mortality rates to newborn screening is that this does not explain why males rather than females would benefit.
There may be several explanations for the delayed reduction in female childhood mortality rates, including differences between the sexes in age of acquisition of Pseudomonas aeruginosa and treatment adherence, hormonal influences or enhanced inflammatory response in females and reluctance to exercise vigorously. Body image issues that may result in nutritional failure as well as differences in resting energy expenditure between males and females may also impact on survival through childhood.25-31 This latter observation may be particularly important, and it is interesting to note that the female group that has experienced no change in mortality events is the vulnerable adolescent group aged 10–14 years. Our overall results for 0–14-year-old Australian children showed more marked reductions in mortality from 1998 onwards among girls than among boys, possibly reflecting the fact that male mortality rates had already fallen substantially and any further improvements were likely to be of much lesser magnitude.
In summary, the pattern of mortality from CF in Australia has changed substantially since 1979. Deaths now occur predominantly in adult years at higher rates for males than for females. Among 0–14 year olds, rates for girls continue to be higher than rates for boys, but mortality in childhood has become uncommon. Survival has increased since 1979, but females continue to have reduced length of life compared with males.
1 Age-standardised rates of deaths and of mortality events (death or lung transplantation) for Australians with cystic fibrosis, 1979–2005
3 Age-specific rates of mortality events (death or lung transplantation) among Australian patients with cystic fibrosis, 1979–1988 and 1989–2005
4 Age-standardised rates of mortality events (age at death in the absence of transplantation, or age at lung transplantation), and female-to-male rate ratios among Australians with cystic fibrosis, 1979–2005
* Number of events. † Per 100 000 population, with Australian census population of 2001 used as the standard. ‡ P < 0.05. |
|||||||||||||||
Competing interests
No relevant disclosures.
Acknowledgements
We thank Professor Trevor Williams, Professor Allan Glanville, Dr Peter Hopkins and Associate Professor Scott Bell for their assistance with collecting deidentified data at the heart and lung transplant centres situated at The Alfred Hospital, Melbourne, St Vincent’s Hospital Sydney and The Prince Charles Hospital, Brisbane, respectively.The Australian Cystic Fibrosis Research Trust provided funding after peer review, but had no involvement in conducting the study, analysing data or writing the manuscript.
References
- Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 2007; 29: 522-526. 0_i1109963
- Rosenfeld M, Davis R, FitzSimmons S, et al. Gender gap in cystic fibrosis mortality. Am J Epidemiol 1997; 145: 794-803. 0_i1109965
- Buchdahl R. Is there a gender gap in cystic fibrosis children? J R Soc Med 2007; 100 Suppl 47: 35-37. 0_pgfId-1155051
- Davis PB. The gender gap in cystic fibrosis survival. J Gend Specif Med 1999; 2: 47-51. 0_pgfId-1155058
- Kulich M, Rosenfeld M, Goss CH, Wilmott R. Improved survival among young patients with cystic fibrosis. J Pediatr 2003; 142: 631-636. 0_i1109969
- Lai HC, Kosorok MR, Laxova A, et al. Delayed diagnosis of US females with cystic fibrosis. Am J Epidemiol 2002; 156: 165-173. 0_pgfId-1155070
- Rawashdeh M, Manal H. Cystic fibrosis in Arabs: a prototype from Jordan. Ann Trop Paediatr 2000; 20: 283-286. 0_pgfId-1155079
- Verma N, Bush A, Buchdahl R. Is there still a gender gap in cystic fibrosis? Chest 2005; 128: 2824-2834. 0_i1109973
- Ketchell RI, Roughton M, Agent P, et al. Predicting survival in end-stage cystic fibrosis. Respir Med 2009; 103: 1441-1447. 0_i1109975
- Ramalle-Gomara E, Perucha M, González MA, et al. Cystic fibrosis mortality trends in Spain among infants and young children: 1981-2004. Eur J Epidemiol 2008; 23: 523-529. 0_i1109977
- Bellis G, et al. Cystic fibrosis mortality trends in France. J Cyst Fibros 2007; 6: 179-186. 0_i1109979
- Halliburton CS, Mannino DM, Olney RS. Cystic fibrosis deaths in the United States from 1979 through 1991. An analysis using multiple-cause mortality data. Arch Pediatr Adolesc Med 1996; 150: 1181-1185. 0_pgfId-1155116
- Fogarty A, Hubbard R, Britton J, et al. International comparison of median age at death from cystic fibrosis. Chest 2000; 117: 1656-1660. 0_i1109982
- Stern M, Wiedemann B, Wenzlaff P; German Cystic Fibrosis Quality Assessment Group. From registry to quality management: the German Cystic Fibrosis Quality Assessment project 1995 2006. Eur Respir J 2008; 31: 29-35. 0_i1109984
- Cystic Fibrosis Foundation. Cystic Fibrosis Foundation, patient registry annual data report 2005. Bethesda, Md: CFF, 2006. 0_i1109986
- United Kingdom Cystic Fibrosis Registry. UK CF Registry annual data report 2008. London: CF Trust, 2009. 0_i1109988
- Singer RB. Cystic fibrosis mortality: registry data of cystic fibrosis. J Insur Med 1997; 29: 233-239. 0_i1109990
- Phelan P, Hey E. Cystic fibrosis mortality in England and Wales and in Victoria, Australia 1976-80. Arch Dis Child 1984; 59: 71-73. 0_i1109992
- Australian Cystic Fibrosis Data Registry. Cystic fibrosis in Australia 2005. Annual report. Sydney: Cystic Fibrosis Australia, 2008. http://www.cysticfibrosis.org.au/pdf/Cystic_Fibrosis_in_Australia_2005.pdf (accessed Jun 2011).
- Nick JA, Chacon CS, Brayshaw SJ. Effects of gender and age of diagnosis on disease progression in long-term cystic fibrosis survivors. Am J Respir Crit Care Med 2010; 182: 614-626. 0_i1109996
- O'Connor GT, Quinton HB, Kahn R, et al; Northern New England Cystic Fibrosis Consortium. Case-mix adjustment for evaluation of mortality in cystic fibrosis. Pediatr Pulmonol 2002; 33: 99-105. 0_i1109998
- Massie RJ, Olsen M, Glazner J, et al. Newborn screening for cystic fibrosis in Victoria: 10 years’ experience (1989–1998). Med J Aust 2000; 172: 584-587. 0_i1110000
- Massie J, Clements B; Australian Paediatric Respiratory Group. Diagnosis of cystic fibrosis after newborn screening: the Australasian experience — twenty years and five million babies later: a consensus statement from the Australasian Paediatric Respiratory Group. Pediatr Pulmonol 2005; 39: 440-446. 0_i1110002
- Grosse SD, Rosenfeld M, Devine OJ, et al. Potential impact of newborn screening for cystic fibrosis on child survival: a systematic review and analysis. J Pediatr 2006; 149: 362-366. 0_i1110004
- Allen JR, McCauley JC, Selby AM, et al. Differences in resting energy expenditure between male and female children with cystic fibrosis. J Pediatr 2003; 142: 15-19. 0_i1110006
- Festini F, Taccetti G, Campana S, et al. Gender differences in the acquisition of P. aeruginosa. Pediatr Pulmonol 2003; 36: 453-454; author reply 454. 0_pgfId-1155224
- Patterson JM, Wall M, Berge J, Milla C. Gender differences in treatment adherence among youth with cystic fibrosis: development of a new questionnaire. J Cyst Fibros 2008; 7: 154-164. 0_pgfId-1155234
- Casimir GJ, Mulier S, Hanssens L, et al. Chronic inflammatory diseases in children are more severe in girls. Shock 2010; 34: 23-26. 0_pgfId-1155241
- Truby H, Paxton AS. Body image and dieting behavior in cystic fibrosis. Pediatrics 2001; 107: E92. 0_pgfId-1155250
- Collins CE, O’Loughlin EV, Henry R. Discrepancies between males and females with cystic fibrosis in dietary intake and pancreatic enzyme use. J Pediatr Gastroenterol Nutr 1998; 26: 258-262. 0_pgfId-1155255
- Zemel BS, Kawchak DA, Cnaan A, et al. Prospective evaluation of resting energy expenditure, nutritional status, pulmonary function, and genotype in children with cystic fibrosis. Pediatr Res 1996; 40: 578-586. 0_i1110013