





Volume 195 - Issue 7
A 17-year-old girl with severe respiratory failure and circulatory shock
Authors: David Gutierrez, Louis Guy, Veera S Katikireddi, Jason P Butler and John Gowardman
Med J Aust 2011; 195 (7): 414-415. || doi: 10.5694/mja11.10238
Published online: 3 October 2011
Published online: 3 October 2011
Clinical record
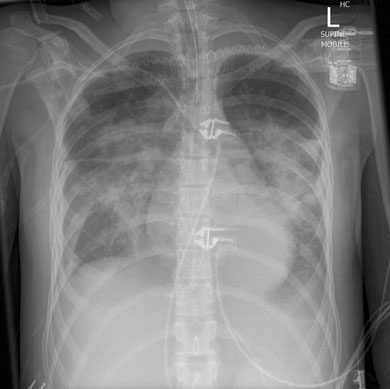
A 17-year-old girl presented to her general practitioner with a 1-week history of fever, arthralgia, general malaise and dry cough. She had a history of systemic onset juvenile idiopathic arthritis (SOJIA), diagnosed at age 2 years and treated with aspirin, and had been in remission for 13 years. She was initially treated by her GP with oral roxithromycin, and admitted to hospital 3 days later with worsening of her symptoms. Her admission chest x-ray (Box 1) revealed bilateral perihilar infiltrates. A diagnosis of severe community-acquired pneumonia was made and broad spectrum antibiotics were commenced, including vancomycin, moxifloxacin, and oseltamivir. Despite this treatment, her condition deteriorated. On Day 3 of admission she required endotracheal intubation and circulatory support with noradrenaline 18–50 μg/kg/min, and was admitted to the intensive care unit (ICU). She remained hypotensive, with a mean arterial pressure of 50 mmHg, and with sinus tachycardia of 132 beats/min, and subsequently required renal replacement therapy. She remained febrile for the first 3 days after admission (temperature range 37.5°C–39°C), and her temperature settled to normal after appropriate therapy was initiated.
Investigations included a computed tomography scan of her abdomen, which showed hepatosplenomegaly, and liver function tests, which showed elevated conjugated bilirubin (36 mmol/L; reference range [RR], < 4 mmol/L), γ-glutamyl transferase (84 U/L; RR, < 24 U/L), lactate dehydrogenase (3540 U/L; RR, 150–280 U/L), alanine aminotransferase (108 U/L; RR, 10–30 U/L) and aspartate transaminase (388 U/L; RR, < 30 U/L). Other abnormal parameters were her haemoglobin level (93 g/L; RR, 120–160 g/L), platelet count (65 x 109/L; RR, 150–400 109/L), white cell count (2.9 x 109/L; RR, 4.5–13 x 109/L), international normalised ratio (2.1; RR, 0.9–1.2) and fibrinogen level (0.9 g/L; RR, > 2.5 g/L). Elevated inflammatory markers included C-reactive protein (301 mg/L; RR, < 5 mg/L) and serum ferritin (50 500 μg/L; RR, 7–140 μg/L). A transthoracic echocardiogram showed a left ventricular ejection fraction of 60%, a mild reduction in right ventricular contractility, and a right ventricular systolic pressure of 48 mmHg. A full screen for sepsis was performed, including a nasopharyngeal aspirate, bronchoalveolar lavage, blood cultures and serological testing; all were unremarkable. Other immunological tests performed were for Mycoplasma pneumoniae antibodies, Streptococcus pneumoniae urinary antigen, Legionella pneumophila urinary antigen, herpes simplex virus, cytomegalovirus, Epstein–Barr virus (EBV) IgM and IgG, influenza A and B, H1N1 influenza RNA, respiratory syncytial virus, parainfluenza and adenovirus DNA, Q fever (Coxiella burnetti) IgM and IgG, and serological tests for hepatitis, dengue fever IgM and Leptospira IgM, all of which were non-reactive. Urinalysis revealed a white blood cell count of 140 x 106/L (RR, < 10 x 106) and an erythrocyte count of > 500 x 106/L (RR, < 10 x 106/L) with no microbial growth on culture. A bone marrow aspirate with trephine was performed on Day 4 (Day 2 in the ICU), and EBV DNA was detected in the resulting sample using a qualitative DNA test.
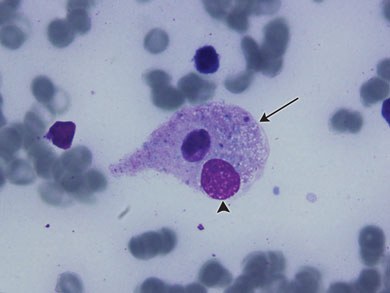
The patient’s failure to improve, in combination with her past history of SOJIA, hepatosplenomegaly and a very high ferritin level led to a preliminary diagnosis of macrophage activation syndrome (MAS). Immunosuppressive treatment in the form of high-dose methylprednisolone (10 mg/kg daily) and intravenous immunoglobulin (1 g/kg daily) were commenced. Inotrope and ventilatory requirements improved within 24 hours of this treatment. On Day 9 of admission she was extubated, and was discharged home on high-dose steroids 8 days later with no complications. Her bone marrow aspirate histological examination showed haemophagocytosis which confirmed the diagnosis of MAS (Box 2).
Macrophage activation syndrome (MAS) is a severe, potentially fatal condition associated with paediatric rheumatic diseases. It is a form of secondary haemophagocytic lymphohistiocytosis (HLH) with uncontrolled activation and proliferation of well differentiated macrophages and T-lymphocytes.1,2 The central pathophysiological abnormality in HLH is cytokine dysfunction, resulting in uncontrolled accumulation of activated T-lymphocytes and activated histiocytes (macrophages) in many organs. High levels of cytokines are found in these patients due to ineffective natural killer cells and T-lymphocytes (a positive feedback loop started by ineffective T-cells, triggering an unopposed release of cytokines that attracts further T-cells).3 Hyperactivated macrophages cause damage in different tissues, and this is thought to be the origin of the high serum ferritin levels characteristic of these conditions. The cause of the macrophage activation is multifactorial. Ineffective cytotoxic immunity is due to underactive natural killer cells and reduced perforin production.
Secondary HLH can be precipitated by infection, drugs, malignancy and rheumatic diseases. SOJIA has been linked to polymorphisms in genes controlling production of cytokines, such as tumour necrosis factor. This could in turn be a cause for MAS, leading some authors to postulate that MAS and SOJIA could be part of the same disease.4
Clinical manifestations include fever (91%–100%), hepatomegaly (90%–92.3%), splenomegaly (77%–84%), lymphadenopathy (41%–62%), neurological symptoms (47%) and rash (43%).5,6 The central nervous system is commonly affected,7 and patients may present with agitation, seizures, coma, respiratory failure from adult respiratory distress syndrome, and multiorgan failure.7 MAS has a mortality of up to 22%.8
To our knowledge, this is the only reported case of MAS in a patient nearing adult age and with a long inactive rheumatic disease period. MAS is well described in the paediatric population with SOJIA, and has a median age of 5 years at the time of presentation.9 The mean time between initial diagnosis of SOJIA and presentation with MAS is 4 years.8 Our patient was aged 17 years at the time of presentation with MAS, and had not had symptoms of, nor required treatment for, SOJIA for 13 years.
Diagnosis was described by Ravelli and colleagues,7 and is based on clinical findings such as organomegaly and laboratory findings such as hypofibrinogenaemia, thrombocytopenia and elevated serum liver enzymes. A bone marrow aspirate can aid diagnosis in uncertain cases, the pathognomonic feature being the presence of well differentiated macrophages actively phagocytosing haemopoietic cells.8 Infections, medications and malignancies have all been identified as triggers for MAS. EBV DNA was found in the patient’s bone marrow aspirate, and EBV has been identified as one of the more common triggers for MAS.5,7 It is likely that EBV was the trigger for this patient’s MAS.
Treatment is based on immunosuppression using steroid therapy. Cyclosporin has been used as the first-line treatment, or used in combination with corticosteroids. Other treatment options include plasma exchanges or intravenous immunoglobulins.10
This patient presented with fever, prominent respiratory failure and cardiovascular collapse. Her initial systemic symptoms and radiological findings suggested severe sepsis, most likely respiratory in origin, her SOJIA had been in remission for over 10 years, and there was no neurological involvement. These factors made clinical suspicion of MAS difficult, but her hepatosplenomegaly and highly raised ferritin levels suggested the diagnosis, which was supported by her laboratory test results (leucopenia, thrombocytopenia, hypofibrinogenaemia and abnormal liver function test results) and bone marrow aspirate histological findings (Box 2).
Despite MAS predominantly presenting in a paediatric population with active disease, this case report emphasises the need to exercise diagnostic vigilance in treating young adult patients with multiorgan failure and a distant history of rheumatic disease.
Lessons from practice
Multiorgan dysfunction and vasodilatory shock may not be of infectious origin.
If a patient fails to improve when treated with broad spectrum antimicrobials, alternative diagnoses should be sought.
Although macrophage activation syndrome (MAS) primarily affects children with active rheumatic disease close to the time of diagnosis, it may occur in adults after a prolonged disease-free period.
Diagnosis of MAS is based on history, clinical examination and laboratory findings. Demonstrating haemophagocytosis in a bone marrow aspirate can aid uncertain diagnosis.
Competing interests
No relevant disclosures.
Acknowledgements
We thank the patient and her family for allowing us to report this case, and Michelle Spanevello for bone marrow aspirate interpretation and photomicrograph editing.
References
- Henter JI, Elinder G, Söder O, et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 1991; 78: 2918-2922. 0_i1139872
- Osugi Y, Hara J, Tagawa S, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood 1997; 89: 4100-4103. 0_i1139874
- Grom AA, Villanueva J, Lee S, et al. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr 2003; 142: 292-296. 0_i1139876
- Félix FH, Leal LK, Fontenele JB. Cloak and dagger: the case for adult onset Still disease and hemophagocytic lymphohistiocytosis. Rheumatol Int 2009; 29: 973-974. 0_i1139878
- Zeng HS, Xiong XY, Wei YD, et al. Macrophage activation syndrome in 13 children with systemic-onset juvenile idiopathic arthritis. World J Pediatr 2008; 4: 97-101. 0_i1139880
- Henter JI, Elinder G, Söder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand 1991; 80: 428-435. 0_i1139882
- Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr 2005; 146: 598-604. 0_i1139885
- Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders Arch Dis Child 2001; 85: 421–426. 0_i1139887
- Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol 2002; 14: 548-552. 0_i1139889
- Buyse S, Teixeira L, Galicier L, et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med 2010; 36: 1695-1702. 0_i1139891