





Volume 194 - Issue 1
An elusive phaeochromocytoma
Authors: Christopher J Yates, Sybil A McAuley, Simon Grodski, Peter Shane Hamblin and Peter R Ebeling
Med J Aust 2011; 194 (1): 44-45. || doi: 10.5694/j.1326-5377.2011.tb04146.x
Published online: 3 January 2011
Published online: 3 January 2011
Clinical record
A 56-year-old woman, with a history of a right adrenal phaeochromo-cytoma excised in 1974, presented to the emergency department in 2008 with a hypertensive crisis characterised by 3 days of severe headache, malignant hypertension (blood pressure, 200/115 mmHg) and seizures complicated by bilateral humeral fractures. After her surgery in 1974, she had unresolved hypertension and raised urinary catecholamine levels suggestive of residual or metastatic disease. However, no additional tumour could be identified despite venous sampling studies. The patient declined further treatment and was lost to follow-up from 1978. She subsequently had three uncomplicated, successful pregnancies.
During the patient’s 2008 hospital admission, her levels of urinary catecholamines and plasma metanephrines were elevated (Box 1). She was treated with phenoxybenzamine and diltiazem (extended release), doses of which were titrated up to 60 mg twice daily and 360 mg daily, respectively, before uneventful surgical repair of both humeral fractures.
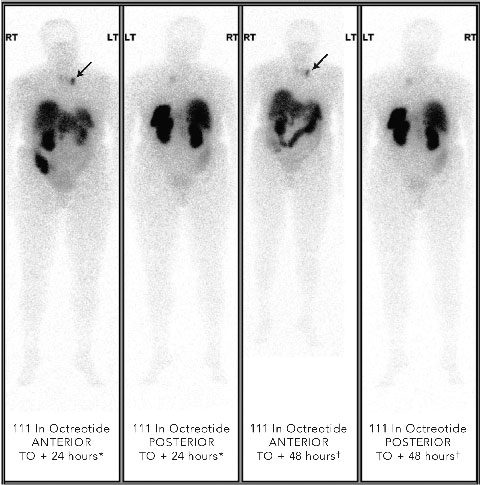
Localisation studies with a 123 I-metaiodobenzylguanidine scan identified increased tracer uptake within both the left adrenal region and left anterior mediastinum. Computed tomography and magnetic resonance imaging (MRI) scans showed large bilateral renal angiomyolipomas (AMLs), a right renal artery aneurysm, a bulky left adrenal gland with no discrete mass lesion, and a 15 mm calcified lesion within the left anterior mediastinum (not related to the sympathetic chain). Results of a positron emission tomography scan were negative. However, an octreotide scan showed mild tracer uptake in the left adrenal region and marked focal uptake within the left anterior mediastinum (Box 2).
It was felt unlikely that the left adrenal gland image represented a phaeochromocytoma and likely that the mediastinal lesion was a phaeo-chromocytoma lymph node metastasis. After the addition of atenolol 100 mg daily to the patient’s drug regimen, the left anterior mediastinal lesion was excised via a cervical approach without complication. Histological examination confirmed a phaeochromo-cytoma metastasis within a lymph node. After the operation, blood pressure improved significantly. Plasma metanephrine levels have remained normal for over a year.
Tests for phaeochromocytoma genetic syndromes revealed a missense mutation (Gly144Arg) on exon 2 of the von Hippel–Lindau (VHL) gene, and we are awaiting testing of the patient’s eight siblings to determine whether or not this is a de novo mutation causing VHL disease. Although two of her three children have inherited the mutation, they have had no disease manifestations. Further investigations of the patient have revealed mild sensorineural hearing loss, but no evidence of haemangio-blastomas via fundoscopy or on MRI scanning of the brain and spinal cord. While there is an increased risk of renal cell carcinoma with VHL disease, recent imaging has shown no change in the patient’s presumed bilateral AMLs and she has declined surgery or embolisation. The left adrenal gland and right renal artery aneurysm have also remained stable on serial imaging. Malignant phaeochromocytoma has been reported to be a risk factor for osteoporosis,1 but the patient’s bone mineral densitometry showed only osteopaenia of her lumbar spine, while her left hip bone measurements were within normal limits.
Our patient’s history shows the need for long-term periodic follow-up of people treated for phaeochromocytomas. It also illustrates the association between von Hippel–Lindau (VHL) disease and phaeochromocytoma. VHL disease, an autosomal dominant condition, is caused by a mutation of the VHL tumour suppressor gene on chromosome 3p; the mutation rate is one in 36 000 live births.2 The VHL gene is involved in regulating the transcription of vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and other hypoxia-inducible proteins. Inactivation promotes tumour angiogenesis and growth through overexpression of VEGF and PDGF receptor agonists, resulting in haemangioblastomas of the central nervous system or retina, pancreatic neuroendocrine tumours and cysts, renal clear cell carcinomas, phaeochromocytoma, endolymphatic sac tumours, and papillary cystadenomas of the epididymis (men) or broad ligament (women).3
In a series report of 246 patients with VHL disease, 26% of patients developed phaeochromocytomas at a mean age of 29 years, 39% had bilateral adrenal phaeochromocytomas, and extra-adrenal disease occurred in up to 30%.4 The adrenal medulla is the main site in the body where the enzyme that converts noradrenaline to adrenaline, phenylethanolamine N-methyltransferase, is localised, and, consequently, adrenal phaeochromocytomas typically have raised levels of both noradrenaline and adrenaline. By contrast, extra-adrenal phaeochromocytomas and phaeochromocytomas in patients with VHL disease do not usually express phenylethanolamine N-methyltransferase and are therefore characterised by raised levels of noradrenaline and normetadrenaline with relatively normal levels of adrenaline and metadrenaline. For our patient, elevations in noradrenaline levels exceeded those of adrenaline both at the time of the original phaeochromocytoma diagnosis and subsequently, with adrenaline levels normalising after the original operation, in keeping with extra-adrenal or metastatic disease. Kindreds with VHL disease can be divided into two types (Types I and II) based upon the incidence of phaeochromocytoma, with Type I generally unaffected and Type II at high risk.5 Type II is further divided — IIA and IIB have a low or high incidence of renal clear cell carcinomas, respectively, while IIC is characterised by the development of phaeochromocytomas without other VHL manifestations. Of the 1548 reported mutations in the VHL gene, 52% are missense mutations and the one previously reported Gly144Arg mutation was associated with polycythaemia.6
Renal angiomyolipomas (AMLs) occur in 0.1% of the population, and although those with bilateral renal AMLs have a greater chance of having tuberous sclerosis, this patient has no other features of the condition.7,8 However, it is possible that she has a new phenotype of Type II VHL disease associated with AMLs.
Between 12% and 24% of patients with ostensibly sporadic phaeochromocytomas harbour phaeochromocytoma genetic syndromes, including VHL disease. A study of 271 such patients detected a germline VHL mutation in 11%, increasing to 42% in those who presented at age 18 years or younger.9 Therefore, it is recommended that patients diagnosed with a phaeochromocytoma before the age of 50 years should undergo screening for underlying germline mutations.10 For relatives identified as having predisposing germline mutations, investigations and follow-up are dictated by the underlying disease, but should include clinical genetics and endocrinological input. In the case of VHL disease, biochemical screening for phaeochromocytomas should start at age 4 years, and tumours should be removed if functional, as indicated by elevated catecholamine levels.4 While our patient’s experience highlights the importance of screening for underlying germline mutations, it also reinforces the unpredictable nature of phaeochromocytoma metastases.
1 Patient’s levels of urinary catecholamines and plasma metanephrines at phaeochromocytoma diagnosis and post-treatment, and metastasis diagnosis and post-treatment
Cr = creatinine. RR = reference range. * Noradrenaline and adrenaline / Cr ratios unavailable. |
|||||||||||||||


2 Octreotide scans of patient with phaeochromocytoma metastasis showing abnormal focal tracer uptake in anterior mediastinum (arrows in anterior views)
 |
|
* Scan 24 hours after intravenous injection of radioactive octreotide. |
Lessons from practice
Persistent hypertension and elevated catecholamine levels after excision of a phaeochromocytoma are suggestive of residual or metastatic disease.
Patients with a history of phaeochromocytoma require long-term surveillance.
Between 12% and 24% of patients with ostensibly sporadic phaeochromocytomas harbour predisposing genetic syndromes, including von Hippel–Lindau (VHL) disease, and genetic screening is recommended in patients diagnosed under 50 years of age.
VHL disease is a dominantly inherited syndrome, involving mutation of the VHL tumour suppressor gene — the mutation occurs in one in 36 000 live births.
Competing interests
None identified.
References
- Glassman AH, Kean JR, Martino JD, Mentser MI. Subtrochanteric pathological fracture of both femora secondary to malignant pheochromocytoma. A case report. J Bone Joint Surg Am 1990; 72: 1554-1558. 0_i1095996
- Lonser RR, Glenn GM, Walther M, et al. von Hippel–Lindau disease. Lancet 2003; 361: 2059-2067. 0_i1095998
- Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol 2004; 22: 4991-5004. 0_i1096000
- Walther MM, Reiter R, Keiser HR, et al. Clinical and genetic characterization of pheochromocytoma in von Hippel–Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol 1999; 162: 659-664. 0_i1096002
- Zbar B, Kishida T, Chen F, et al. Germline mutations in the von Hippel–Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat 1996; 8: 348-357. 0_i1096004
- Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, et al. Genetic analysis of von Hippel–Lindau disease. Hum Mutat 2010; 31: 521-537. 0_i1096006
- Fujii Y, Ajima J, Oka K, et al. Benign renal tumors detected among healthy adults by abdominal ultrasonography. Eur Urol 1995; 27: 124-127. 0_i1096008
- van Baal JG, Smits NJ, Keeman JN, et al. The evolution of renal AMLs in patients with tuberous sclerosis. J Urol 1994; 152: 35-38. 0_i1096010
- Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346: 1459-1466. 0_i1096012
- Pigny P, Cardot-Bauters C, Do Cao C, et al. Should genetic testing be performed in each patient with sporadic pheochromocytoma at presentation? Eur J Endocrinol 2009; 160: 227-231. 0_i1096016