





Volume 193 - Issue 10
Twenty-five years of treatment for childhood acute lymphoblastic leukaemia in Western Australia: how do we compare?
Authors: Hannah Forward, Guicheng C Zheng and Catherine H Cole
Med J Aust 2010; 193 (10): 585-589. || doi: 10.5694/j.1326-5377.2010.tb04067.x
Published online: 15 November 2010
Published online: 15 November 2010
Abstract
Objectives: To compare survival among the subgroup of children with acute lymphoblastic leukaemia (ALL) who were treated at Princess Margaret Hospital for Children (PMH) in Perth, Western Australia, over 25 years under 15 consecutive protocols of the Children’s Cancer Group (CCG) with survival for the entire cohort of children in multiple centres treated under CCG protocols in that period; and to highlight the benefits of membership of a large cooperative research group conducting multicentre randomised controlled trials.
Design, participants and setting: Retrospective review of the outcomes of all 311 children with newly diagnosed ALL treated at PMH between 1983 and 2008.
Main outcome measures: 4-year event-free survival; and 10-year overall survival.
Results: Four-year event-free survival for the entire PMH cohort increased from 66% (SE, 6%) for 1983–1987 to 88% (SE, 6%) for 2002–2005, while overall survival over the same period improved from 78% (SE, 5%) to 94% (SE, 4%). Comparisons of outcomes of children treated at PMH with those of the entire CCG cohort, protocol by protocol, revealed similar outcomes.
Conclusion: Outcomes of children treated at PMH over the 25-year period are equivalent to those of the larger CCG cohort.
Between 1983 and 2008, all Western Australian children with newly diagnosed acute lymphoblastic leukaemia (ALL) were treated at Perth’s Princess Margaret Hospital for Children (PMH) under 15 consecutive protocols from trials conducted by the North American Children’s Cancer Group (CCG).
The CCG (now part of the Children’s Oncology Group) was a cooperative research group of more than 120 centres in the United States, Canada and Australia. More than 16 000 children have been treated in CCG randomised controlled trials for ALL since 1968.1 The event-free survival (EFS) of children with ALL has risen from 15% to over 80% during the past four decades. This can be attributed to the intensification of therapy with agents previously shown to be effective in the treatment of ALL in a series of randomised controlled trials performed by the dozen or so international, multicentre, cooperative groups in high-income countries around the world. EFS is predicted based on clinical and biological or disease variables, and treatment intensity is modified according to EFS to maximise cure while minimising toxicity. It is notable that few new drugs have contributed to this improvement in outcome (rather, there has been better use of existing drugs and improved supportive care) and that the CCG studies are not sponsored by the pharmaceutical industry.
In 1983, the Total Care Unit for Children with Cancer at PMH became an associate member of the CCG and began to treat patients with ALL according to CCG protocols. By 1992 the unit had fulfilled the requirements for compliance and audit, and was accepted as the first non-North American full member of the CCG enrolling patients in the study.
The last CCG ALL protocol (CCG 1991) closed to accrual in January 2005, and by early 2008, all patients had completed their therapy and were in follow-up. In 2000, the CCG joined with the other large cooperative group for the management of children with cancer and leukaemia in North America, the Pediatric Oncology Group, and formed the Children’s Oncology Group (COG). Randomised trials of treatment for children with ALL continue within the COG, but there are as yet no mature data for analysis.
The incidence of childhood ALL in WA is similar to that in other Western populations — 3.7 per 100 000 person-years.2 Despite being the commonest childhood malignancy, only 15–20 new patients aged under 16 years are diagnosed with ALL in WA each year. This low incidence makes meaningful, Phase III, randomised controlled trials of treatment impossible, and highlights the importance of membership of a large cooperative clinical trials group so that many hundreds of patients can be studied within a reasonable time frame. Without membership of CCG, the small cohort of patients in WA may have been treated according to the best available evidence, such as that of the most recently published peer-reviewed trials of therapy. Even within the large cooperative groups, the time taken from opening a Phase III randomised clinical trial for ALL to the publication of a manuscript is about 10 years. Thus children treated according to the latest published research would receive therapy about 10 years out of date compared with children treated in the clinical trial.
However, as well as reporting overall improvements in outcomes of CCG patients with successive trials of therapy, it is important for us to review the outcomes of our Western Australian patients separately, to ensure that they are achieving equivalent success. As we contribute only 1%–2% of patients in the entire CCG cohort, it is possible that our patients are faring comparatively badly within the larger cohort.
In this study, we aimed to review the outcomes of all children who began treatment at PMH between 1983 and 2005 to determine survival according to treatment protocol, and to compare this with the published results for the entire CCG cohort.
Methods
For the period 1983 to 2008, records of children aged 0–16 years with ALL were reviewed retrospectively with respect to date of diagnosis, treatment protocol and outcome. Fifteen protocols were used in treatment. Each successive ALL study accrued patients for about 5 years.
Patients diagnosed before 1992 were treated according to the best arm of the previous closed study — the standard arm of the current open protocol. After 1992, 212 children were treated at PMH, of whom 146 (69%) consented to be enrolled in the current randomised study and 66 (31%) were treated off-study on the standard arm. Reasons for children not being enrolled in the study included lack of an available study open at the time of diagnosis, patient ineligibility (eg, prior treatment with corticosteroids) and parents’ decisions for reasons such as a perceived increased burden of therapy on some experimental arms.
Long-term follow-up was assessed in the Total Care Unit’s “late effects” clinics from 5 years after diagnosis and, after about 10 years in remission, by annual postal questionnaire.
Treatment
Risk-adjusted therapy is the primary principle underpinning the treatment of childhood ALL. Treatment allocation is based on risk of relapse determined by presenting features such as age, total white blood cell count (WCC) at diagnosis, leukaemia immunophenotype and cytogenetics, and early response to therapy measured by disappearance of peripheral blood leukaemic blasts, microscopic marrow response (remission), and/or laboratory measurement of submicroscopic leukaemia, termed minimal residual disease.3
CCG treatment protocols
100 series: The CCG-100 series of risk-adjusted protocols was used between 1983 and 1989, based on new treatment strategies to intensify therapy for all risk groups and reduce the need for prophylactic cranial irradiation in selected patients. Patients were risk-stratified according to clinically available prognostic factors including age, WCC, sex, platelet count, and presence of lymphomatous features.1 The crucial contributions of the CCG-100 series were identification of the benefit of intensification of therapy (delayed intensification) and that prophylactic cranial irradiation was not needed for lower-risk and intermediate-risk patients provided they received intensified systemic therapy.4
1800 series: Between 1989 and 1995, 5147 patients were enrolled in the CCG studies for ALL, and 89 children at PMH were treated according to CCG-1800 series protocols. Patients were stratified by age, WCC, sex, platelet count, and lymphomatous features.
Lower-risk patients with rapid marrow remission were randomly allocated to receive the addition of a single delayed intensification (DI) phase of therapy. This was well tolerated and increased 7-year remission rates by 6%.5 Subsequently, standard-risk patients were randomly allocated to receive either dexamethasone or prednisone in induction and maintenance. The results showed that the use of dexamethasone resulted in a significant improvement in EFS compared with prednisone.6
For intermediate-risk patients, a second DI (DDI) phase improved outcome over a single DI phase using modified Berlin–Frankfurt–Münster therapy that included prednisone, particularly for subsets of patients who showed delayed early responses to induction therapy.7,8
Postinduction intensification (increased dose intensities and prolonged duration of therapy) resulted in greater toxicities, but significantly improved the outcomes of high-risk children.9 However, patients with lymphomatous features did not benefit from the use of prophylactic granulocyte colony-stimulating factor.10
In the CCG-1800 series period, infants with ALL continued to represent a very high-risk group. The 4-year EFS remained at only 39%, which was a relatively small improvement from the previous infant trial, which had a 4-year EFS of 33%. Later studies tested the hypothesis that intensifying early therapy for infants would improve outcomes.11
1900 series: The CCG-1900 series was conducted between 1995 and 2002. Intrathecal triple therapy (methotrexate, cytosine arabinoside and hydrocortisone) decreased central nervous system relapse in standard-risk patients, but failed to improve overall outcome when compared with intrathecal methotrexate alone.12
High-risk children who had a rapid marrow response to induction therapy did better with more intensive postinduction therapy, while prolonged duration of therapy added no further benefit.13
The final CCG study for standard-risk ALL patients concluded that there were no benefits to DDI over DI in children with a favourable early marrow response to induction chemotherapy, but that intravenous methotrexate improved outcomes compared with oral methotrexate.14,15
Furthermore, we can now define, by the cytogenetics of the leukaemia cells, a low-risk group of patients almost certain to be cured with graduated intensity therapy.
Statistical analysis
We analysed patient information current to 1 January 2008. Outcome analyses used lifetable methods, Kaplan–Meier procedures and associated statistics. The primary end-point examined was EFS from the time of entry into the study. Events used to calculate EFS were defined as induction failure or death, leukaemic relapse at any site, death during remission or second malignant neoplasm — whichever occurred first. Patients without events at EFS analysis were censored at the date of last contact.
To determine if the PMH outcomes were equivalent to the CCG outcomes, the hypothesis we tested was that the percentage EFS for the CCG was the same as that for PMH (ie, Ho: EFS[CCG] = EFS[PMH]). This was tested using a two-sided P value for the difference between the two proportions using a normal approximation. Statistically significant difference was defined as P < 0.05.
Results
The entire cohort of children treated at PMH for ALL under CCG protocols from 1983 to 2008 numbered 311; 133 were female (43%) and 178 were male (57%). There were 85 events among the overall cohort: 72 relapses, 11 deaths in remission (some following allogeneic haemopoietic stem-cell transplantation) and two diagnoses of a second malignant neoplasm (one of chronic myelomonocytic leukaemia and the other of malignant astrocytoma).
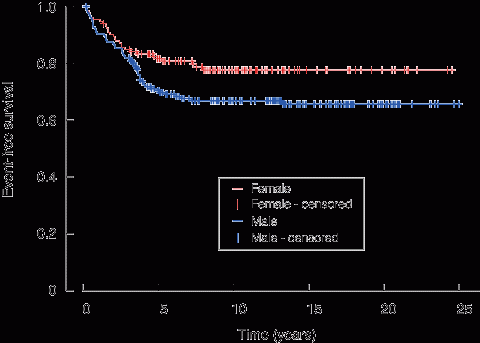
Box 1 shows that the PMH cohort had a superior outcome for female patients, consistent with that observed in other CCG studies.1
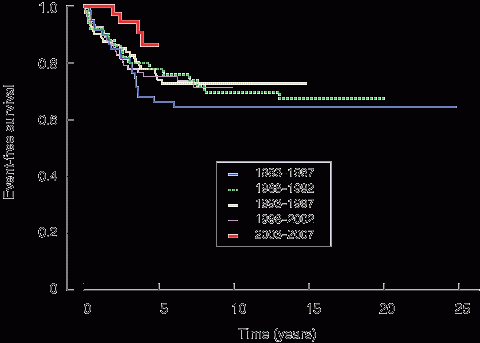
Four-year EFS for the entire PMH cohort increased from 66% (SE, 6%) for 1983–1987 to 88% (SE, 6%) for 2002–2005, while overall survival over the same period improved from 78% (SE, 5%) to 94% (SE, 4%). EFS by period is illustrated in Box 2.
CCG-100 series
The median duration of follow-up for both the PMH cohort and overall CCG cohort was 12 years.4 In aggregate, the overall survival on the CCG-100 protocols at 10 years for the PMH cohort was 79% (SE, 5%) compared with 73% for the CCG cohort.
Results of individual studies for children treated on the five protocols of the CCG-100 series are summarised in Box 3. At PMH, only two infants were treated, and both relapsed within a year. This sample size is too small to calculate lifetable values, so EFS for infants treated on protocol 107 at PMH cannot be estimated.
For most of the protocols, the null hypothesis was not rejected at the 5% level of significance, and hence no significant difference was found between the EFS rates of the CCG overall and PMH. However, the PMH 10-year overall survival for children treated on CCG protocol 105 was 88%, while the equivalent CCG overall survival was only 78%. This difference in outcome was statistically significant (P = 0.04).
CCG-1800 and CCG-1900 series
Results of individual studies for children treated on the 10 protocols of the CCG-1800 and CCG-1900 series are summarised in Box 4.
Only three infants were treated at PMH under a CCG-1800 series protocol, and all relapsed and died within a year of diagnosis. This sample size is too small to calculate lifetable values, so neither EFS nor overall survival for infants treated on protocol 1883 at PMH can be estimated.
For all of the remaining CCG-1800 and CCG-1900 series protocols, the null hypothesis was not rejected at the 5% level of significance, and hence no significant difference was found between the EFS rates of the CCG cohort overall and the PMH cohort treated on these protocols.
Discussion
The EFS of Western Australian children with ALL treated at PMH over 25 years according to CCG protocols are equivalent to the outcomes of the larger CCG cohort. These results reassure us that we are delivering effective anti-leukaemic therapy and supportive care to our patients with ALL.
As PMH is the only paediatric hospital in WA, all newly diagnosed patients with ALL are referred to the Total Care Unit for Children with Cancer for treatment. The small population of WA coupled with the relatively low incidence of ALL result in very small numbers of patients on any treatment protocol.
Our retrospective study has some limitations. Children treated “on study” at PMH after 1992 are also included in the CCG survival data. However, as they make up less than 2% of the entire CCG cohort, this bias was considered negligible. Second, our analysis was unable to examine prognostic factors of the PMH cohort within each treatment protocol group, and the groups may not be equivalent for important prognostic factors such as age or WCC. This may be the reason for a better outcome for PMH patients treated according to the intermediate-risk protocol CCG 105. Before 1992, PMH patients were treated on the standard arm of the current protocol and in all but one CCG ALL study, the experimental arm was the better arm of the study. We may, therefore, have expected the outcome for PMH patients to have been poorer than for the CCG as a whole for these studies, yet no definite trend was seen. The differences in outcome between PMH and the whole CCG cohort for each remaining study are neither statistically nor clinically significant.
The small numbers of patients in each risk-stratified treatment group of PMH patients and the large standard errors associated with the survival data in such small cohorts highlight the statistical power of large cohorts when treatment centres work cooperatively, as in the CCG. Only with large numbers of patients is there enough statistical power for randomisation between standard and experimental treatments to show a difference in outcome within a reasonable time frame. The CCG study cohorts are of adequate sample size to detect moderate treatment differences of the order of a 25%–30% reduction in hazard, even when the difference in EFS between arms is only a small percentage.1 Such a sample size ensures that useful interventions that boost EFS are identified and are used to direct future treatment.
The national and international cooperation in obtaining such results has been an enormous feat of organisation. All children with cancer in Australia are now offered the opportunity to take part in clinical trials from North America and Europe. Our continued cooperation with other international centres is essential to continue to improve survival for these children. Barriers to participating in international multicentre cooperative group trials include regulatory hurdles (which prevent Australian centres from benefiting from the centralised ethics submission process available to US centres), transport costs and time delays in shipping specimens to central laboratories for testing.
Finally, while there is no doubt that paediatric patients with cancer and leukaemia are best treated in large, randomised, international, cooperative group trials, the lack of government funding for data management and the lack of individual academic recognition for such participation may lead clinical researchers to pursue smaller trials, perhaps to the detriment of Australian patients.16
1 Event-free survival of children treated at Princess Margaret Hospital on Children’s Cancer Group protocols, 1983–2008
2 Event-free survival of patients treated at Princess Margaret Hospital on Children’s Cancer Group protocols during five periods, 1983–2007
3 Event-free survival and overall survival data for Children’s Cancer Group (CCG)-100 series compared with the Princess Margaret Hospital (PMH) cohort
SE = standard error. |
|||||||||||||||
4 Event-free survival for the Children’s Cancer Group (CCG)-1800 series protocols onwards: comparison of the overall CCG cohort and the Princess Margaret Hospital (PMH) cohort
SE = standard error. |
|||||||||||||||
Competing interests
None identified.
Acknowledgements
We acknowledge Michael Willoughby and David Baker, Children’s Cancer Group principal investigators at Princess Margaret Hospital during these Children’s Cancer Group studies. We also acknowledge that Professor M K Bulsara and Professor K Hird of the School of Medicine, University of Notre Dame, Fremantle, WA contributed to the design and analysis of this study.
References
- Gaynon PS, Trigg ME, Heerema NA, et al. Children’s Cancer Group trials in childhood acute lymphoblastic leukemia: 1983–1995. Leukemia 2000; 14: 2223-2233. 0_i1095973
- Milne E, Laurvick CL, de Klerk N. Trends in childhood acute lymphoblastic leukemia in Western Australia, 1960–2006. Int J Cancer 2008; 122: 1130-1134. 0_i1095975
- Schultz KR, Pullen DJ, Sather HN, et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG). Blood 2007; 109: 926-935. 0_i1095977
- Trigg ME, Sather HN, Reaman GH, et al. Ten-year survival of children with acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Leuk Lymphoma 2008; 49: 1142-1154. 0_i1095979
- Hutchinson RJ, Gaynon PS, Sather HN, et al. Intensification of therapy for children with lower risk acute lymphoblastic leukemia: long-term follow-up of patients treated on Children’s Cancer Group Trial 1881. J Clin Oncol 2003; 21: 1790-1797. 0_i1095981
- Bostrom BC, Sensel MR, Sather HN, et al. Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Blood 2003; 101: 3809-3817. 0_i1095983
- Lange BJ, Bostrom BC, Cherlow JM, et al. Double-delayed intensification improves event-free survival for children with intermediate-risk acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Blood 2002; 99: 825-833. 0_i1095985
- Riehm H, Feickert HJ, Shrappe M, et al. Therapy results in five ALL-BFM studies since 1970: implications for risk factors for prognosis. Hamatol Bluttransfus 1987; 30: 139-146. 0_i1095987
- Nachman JB, Sather HN, Sensel MG, et al. Augmented post induction therapy for children with high risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med 1998; 338: 1663-1671. 0_i1095989
- Heath JA, Steinherz PG, Altman A, et al. Human granulocyte colony-stimulating factor in children with high-risk acute lymphoblastic leukemia: a Children’s Cancer Group Study. J Clin Oncol 2003; 21: 1612-1617. 0_i1095991
- Reaman GH, Sposto R, Sensel MG, et al. Treatment outcome and prognostic factors for infants with acute lymphoblastic leukemia treated on two consecutive trials of the Children’s Cancer Group. J Clin Oncol 1999; 17: 445-455. 0_i1095993
- Matloub Y, Lindemulder S, Gaynon PS. Intrathecal triple therapy decreases central nervous system relapse but fails to improve event-free survival when compared with intrathecal methotrexate: results of the Children’s Cancer Group (CCG) 1952 study for standard-risk acute lymphoblastic leukemia, reported by the Children’s Oncology Group. Blood 2006; 108: 1165-1173. 0_i1095995
- Seibel NL, Steinherz PG, Sather HN, et al. Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood 2008; 111: 2548-2555. 0_i1095997
- Matloub Y, Angiolillo A, Bostrom B, et al. Double delayed intensification (DDI) is equivalent to single DI (SDI) in children with National Cancer Institute (NCI) standard-risk acute lymphoblastic leukemia (SR-ALL) treated on Children’s Cancer Group (CCG) clinical trial 1991 (CCG-1991) [abstract]. Blood 2006; 108: A-146. 0_i1095999
- Matloub Y, Bostrom BC, Angiolillo AL, et al. Children with NCI standard risk acute lymphoblastic leukemia and TEL-AML1 or favorable chromosome trisomies are almost certain to be cured with graduated intensity therapy: results of the CCG 1991 study. Blood 2010. In press. Epub ahead of print 7 Dec 2009. http://ash.confex.com/ash/2009/webprogram/Paper 20261.html (accessed Aug 2010).
- Booth CM, Tannock I. Reflections on medical oncology: 25 years of clinical trials — where have we come and where are we going? J Clin Oncol 2008; 26: 6-8. 0_i1096004