




The term carcinoid (carcinoma-like) was introduced by Oberndorfer in 1907 to describe a tumour of the gastrointestinal tract that was less aggressive than adenocarcinoma.1 The earliest clear description of carcinoid syndrome (flushing, diarrhoea, and bronchospasm) and carcinoid heart disease was published in the early 1950s.1 At the same time, the first systemic biogenic amine producing these symptoms, 5-hydroxytryptamine, was identified,1 and the neuroendocrine origin of carcinoid tumours was established.
Carcinoid tumours were originally classified according to their location (foregut, midgut or hindgut). The current recommended classification system, which is more broadly applicable, was developed by the World Health Organization and published in 2000.2 This taxonomy uses the generic term neuroendocrine tumour (NET), and tumours are classified into one of four types based on their size, proliferative rate, localisation, differentiation, and hormone production:
Type 1: Well differentiated NET (benign behaviour);
Type 2: Well differentiated NET (uncertain behaviour);
Type 3: Well differentiated neuroendocrine carcinoma (NEC) (low-grade malignancy); and
Type 4: Poorly differentiated NEC (high-grade malignancy).
Recent reports have emphasised the value of the WHO classification and the tumour–node–metastasis staging system for determining prognosis for both gastrointestinal luminal and pancreatic NETs.3,4 In our article, we use “NETs” to include all the four tumour types in the WHO classification.
As all NETs have the potential to produce bioactive amines that can cause dramatic systemic symptoms, “functioning” tumours may be discovered when they are quite small. Clinically “silent” or “non-functioning” tumours may produce local symptoms as they grow, or when they metastasise to the liver. Local symptoms include obstruction (bile duct or bowel), perforation, or bleeding in the gastrointestinal tract. Conversely, asymptomatic NETs may be discovered incidentally; for example, in the pancreas during imaging of the abdomen, or in the stomach or colon during endoscopic screening for other conditions. Based on the varying neuroendocrine cell types involved in the genesis of NETs, and the subsequent diversity of the secretory spectrum of peptides and amines produced, patients with gastrointestinal NETs present with subtle and protean clinical symptoms. This can lead to a delay in diagnosis of up to 5–7 years,5 or result in inappropriate management.
National and international efforts are now underway to develop practice guidelines for diagnosing and treating this heterogeneous disease, and to establish multidisciplinary management groups.6 In Australia, the number of gastrointestinal NETs treated on a per capita basis is far lower than the predicted incidence (1500–2000 new cases per year — United States National Cancer Institute, Surveillance Epidemiology and End Results database),7,8 suggesting significant under-recognition of a disease entity whose predicted prevalence is 5000–7000 cases.7,8
The frequency of NETs in large autopsy series, before the era of increased detection by computed tomography (CT) and endoscopy, indicated that about 85% were undiagnosed during life.9 US data show that the incidence of gastrointestinal NETs has increased at a rate of 3%–10% per year over the past three decades.7,8 This most likely reflects:
increased detection — greater use of abdominal CT and endoscopy, and advances in nuclear medicine and immunohistochemical pathology leading to improved diagnostic classification;
increased awareness of the disease among physicians; and
a true increase in tumour incidence.
Similar trends are evident from European cancer registries.
The most common site of primary NETs is the gastrointestinal tract (about 60% of all cases), followed by the bronchopulmonary tree (27%). Less frequent sites are the ovaries, testes, liver, biliary tract and pancreas.10 Within the gastrointestinal tract, the most common site is the small intestine (34%), followed by the rectum (23%), colon (19%), stomach (7.7%), pancreas (7.5%) and appendix (6.6%). As a result of the substantial (~ 60%) 5-year survival, the prevalence of NETs (about 35 per 100 000) is considerably higher than the incidence and, as a group, NETs are more prevalent than either gastric, pancreatic, oesophageal or hepatobiliary adenocarcinomas, or any two of these cancers combined.7
Most gastrointestinal NETs can be recognised readily by routine histological examination, but immunohistochemical analysis may be necessary for confirmation. The most useful marker of neuroendocrine cells in tissue sections is chromogranin A (CgA), a glycoprotein stored in secretory granules of neuroendocrine cells. In occasional instances, synaptophysin immunostaining, a neuron-specific enolase test, or tests for specific peptide hormone markers, such as serotonin, somatostatin and gastrin, may be useful.11 Further, establishing the proportion of proliferating cells (mitotic count and Ki-67 index [a marker of cell proliferation]) within the tumour is important, as it provides some indication of prognosis.
The lack of global uniformity of nomenclature, classification and grading of these tumours has impeded progress in understanding their biology. Publication of the recent US consensus document on the pathology of NETs provides clarification and a minimum dataset for uniform pathological evaluation.12
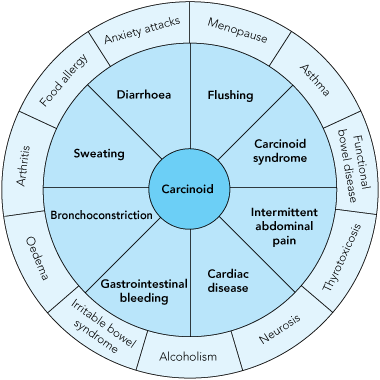
As previously stated, erroneous or delayed diagnosis is common, as most gastrointestinal NETs are usually small (< 2 cm) at presentation, initially asymptomatic, or the symptoms are misdiagnosed as more prosaic or mundane entities (eg, food allergy, anxiety disorders, menopause symptoms or irritable bowel syndrome; Box 1). The bioactivity of the individual amines and peptides secreted (serotonin, catecholamines, dopamine, histamine, gastrin, glucagon, and prostaglandins, among others) may have symptomatic correlates. The classical carcinoid syndrome is relatively uncommon (10%–15% of diagnosed cases), and typically consists of diarrhoea, cutaneous flushing, bronchospasm and right-sided heart failure (the latter due to concurrent right-sided cardiac valve fibrosis caused by serotonin).10,14 The carcinoid syndrome (or symptoms from secreted bioactive amines) usually occurs after the tumour metastasises.
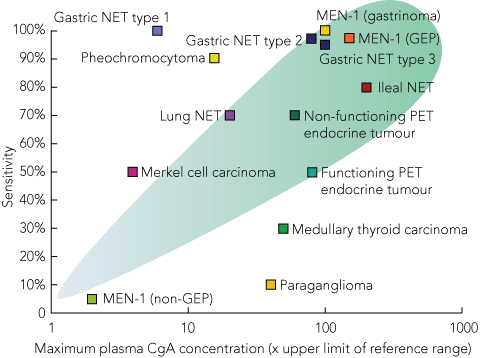
Measurement of plasma CgA concentration is the most useful diagnostic test for all NETs (Box 2). Although the plasma CgA level is a very sensitive marker of NETs,15 CgA levels can also be elevated in other conditions, including pancreatic and small-cell lung cancer and some prostate carcinomas;16 in renal impairment and atrophic gastritis; and particularly during proton-pump inhibitor therapy.17 Plasma CgA levels correlate reasonably well with tumour burden, and may be useful for monitoring treatment.18
Twenty-four-hour measurement of urinary 5-hydroxyindole-3-acetic acid (5-HIAA), the degradation product of serotonin, is a useful laboratory marker for serotonin-producing NETs, but the assay is a complex one. The specificity of 5-HIAA as a marker for these NETs is 88%, although tryptophan/serotonin-rich foods (bananas, avocados, plums, eggplants, tomatoes, plantains, pineapples and walnuts) can provide false elevations, and several drugs can result in increased or decreased 5-HIAA levels.13 Higher concentrations of 5-HIAA in urine are consistent with a worse prognosis,18 while persistently low levels predict a more favourable outcome in disseminated disease.19
Upper endoscopy can detect oesophagogastric lesions, and colonoscopy can detect colorectal and some terminal ileal tumours. For the rest of the small intestine, double balloon or push enteroscopy is reasonably effective, although time-consuming and uncomfortable for the patient.20 Capsule endoscopy (in which a video capsule is swallowed to photograph the oesophagus, stomach, and small intestine) is more patient-friendly but lacks biopsy capabilities.21
Endoscopic ultrasound examination is highly sensitive for detecting NETs of the stomach, duodenum, pancreas and rectum, as it identifies submucosal lesions and facilitates staging. Endoscopic ultrasound-guided, fine-needle aspiration is particularly useful for diagnosing pancreatic NETs.22 Conventional ultrasound examination can be useful intraoperatively, but is relatively poor at imaging liver metastases of NETs. Contrast-enhanced ultrasound gives a better image, improving detection rates from 68% to 98%.23
Enteroclysis and barium-contrast studies have been widely supplanted by CT and magnetic resonance imaging (MRI).24 Small primary tumours are difficult to visualise unless there is secondary fibrosis. Characteristic findings include mass lesions, radiating strands of fibrosis and spiculation (calcification) with traction or fixation of bowel.
Radiological examination is useful in the initial localisation of gastrointestinal NETs, but nuclear imaging using tumour-specific radiolabelled receptor analogues, such as 111In-octreotide, is considerably more sensitive and specific for detecting NET disease. Five somatostatin receptor subtypes have been identified. About 70%–90% of gastrointestinal NETs express multiple receptor subtypes (predominantly subtypes 2 and 5), such that about 90% of NETs can be detected with radiolabelled somatostatin analogues (SSAs).13 Somatostatin receptor scintigraphy, particularly with single-photon emission CT (SPECT), is effective in monitoring the efficacy of therapy and assessing disease progression. The amount of radionuclide uptake can also help to predict the potential therapeutic effect of SSAs (see discussion of SSAs under the heading Biotherapy).25 Inflammatory conditions, including Crohn’s disease, can produce false-positive results, as cells of the inflammatory response express somatostatin receptors.26 Hybrid SPECT examination can better localise disease deposits and differentiate lesions from physiological uptake, which improves diagnostic accuracy.
Positron emission tomography (PET) using 11C-labelled 5-hydroxytryptophan is also used for imaging. Conventional PET scanning with 18F-fluorodeoxyglucose is rarely helpful, as most NETs have low glycolytic rates. Alternative markers are under investigation. PET using gallium-labelled octreotide derivatives will probably replace octreotide somatostatin receptor scintigraphy in the near future, because it is cheaper and easier to perform, and has a higher sensitivity.27
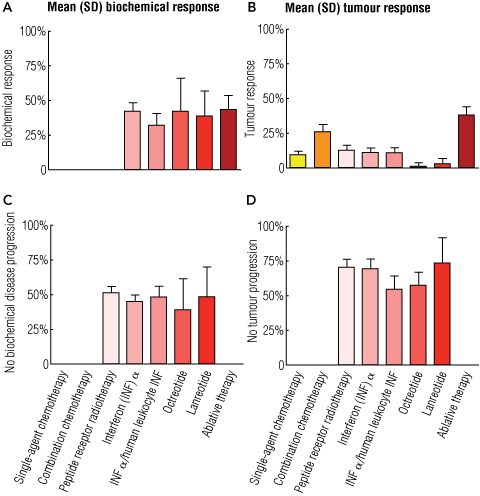
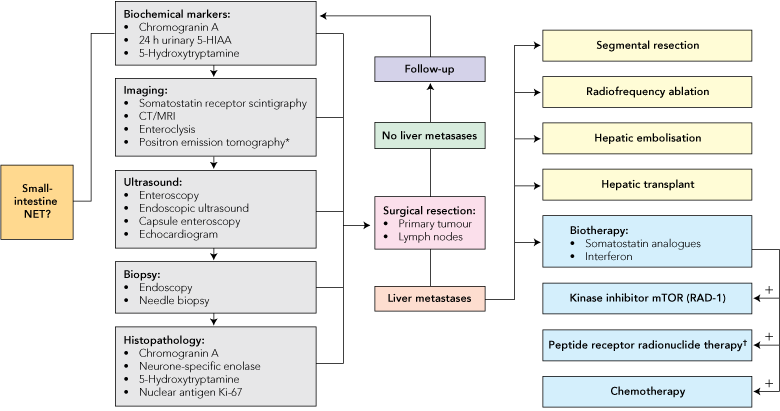
The management of gastrointestinal NETs depends on many factors, including operability; symptoms; tumour type, stage and grade; and the patient’s overall medical condition. The primary site of the tumour also determines management strategy, as highlighted in Box 3. Most therapeutic decisions are currently based on empirical evidence, as properly designed trials for this heterogeneous condition are lacking.31 Summary data on treatment efficacy are presented in Box 4, and an overall strategy for management of suspected small-intestine NETs is given in Box 5. As there are no cellular or biological differences between functioning NETs (which secrete peptides and produce hormonal syndromes) and non-functioning NETs, their treatment is based on the same principles.
Primary surgical resection of the tumour and regional lymph nodes is the only curative treatment for gastrointestinal NETs; this is usually possible in about 20% of patients. Small, solitary non-invasive, endosonographically proven lesions in the stomach, duodenum and rectum may be resected endoscopically. Surgical intervention alone or in combination with other treatment options is appropriate palliation for incompletely resectable disease, as it increases median survival, decreases tumour burden, facilitates symptom control, and decreases local and systemic complications.32 Cardiorespiratory consequences can occur when bioactive agents are released during the procedure, so patients should be given SSAs intravenously during anaesthesia to obviate this.33
Somatostatin analogues: Somatostatin is a neurotransmitter peptide that, in general, inhibits cellular secretion. Pharmacological reconfiguration of native somatostatin, a tetradecapeptide, to an octapeptide (octreotide) substantially prolongs the half-life of the peptide, thus improving its efficacy. The currently available SSAs — Sandostatin LAR (octreotide; Novartis) and Somatuline Autogel (lanreotide; Ipsen) — display high-affinity binding for receptor subtypes 2 and 5, low affinity for subtypes 1 and 4, and medium affinity for subtype 3. SOM230 (pasireotide; Novartis), a more recently developed SSA, now in Phase III trials, has a wider range of activity against somatostatin receptors and may offer a therapeutic advantage, especially in resistant disease.34 SSAs are generally well tolerated and offer the best therapeutic option for inducing biochemical responses and managing clinical symptoms in patients with gastrointestinal NETs. These long-lasting depot formulations (Sandostatin LAR and Somatuline Autogel) are administered once a month, largely eliminating the need for daily subcutaneous injections.
There is some controversy about whether SSA therapy is antiproliferative, as tumour size rarely decreases (Box 4). Recent data, however, provide evidence that long-term administration of Sandostatin LAR inhibits tumour growth and prolongs progression-free survival in patients with well differentiated NETs of the midgut.35 General opinion favours treating both functioning and non-functioning NETs with SSAs, regardless of whether or not the tumours produce a distinct clinical syndrome.
Other biotherapies: Therapies based on inhibition of growth factors or their receptors, or downstream signal transduction pathways, have been used recently. Responses have been modest when single-agents have been used, although a combination of everolimus (mTOR kinase inhibitor) and Sandostatin LAR has provided initially encouraging results.5
Chemotherapy: Response rates for single-agent conventional chemotherapeutic agents are generally low, of the order of 5%–10%.5 Although combination chemotherapy may give slightly better response rates (20%–30%), the results remain disappointing and adverse effects are often substantial. Use of chemotherapy is thus usually confined to patients with more aggressive tumours, indicated by high mitotic rates or Ki-67 indices.
In contrast to the relative ineffectiveness of radiation by external beam, peptide receptor radionuclide therapy delivers tumoricidal doses of radiation to NET cells highly selectively, with few adverse effects (nausea and occasional bone marrow and renal toxicity). By linking a radioactive isotope (111Indium, 90Yttrium or 177Lutetium) to an SSA, NET cells, with their often high density of somatostatin receptors, may be specifically targeted. Individual isotopes have advantages and disadvantages as regards the types of radiation emitted. This therapy is dependent on a high SSA uptake by tumour cells, and its success can be predicted to some extent by somatostatin receptor scintigraphy.36 Tumour regression rates of up to 50%, with a disease-free response approaching 3 years, have been reported in some studies.36
Unfortunately, most patients have multiple, bilateral liver metastases at the time of diagnosis. Options include lesion ablation (by radiofrequency, cryoablation or embolisation) as well as surgical resection — ranging from cytoreductive (excisional) surgery to liver transplantation. Improved 5-year survival has been reported after resection of hepatic metastases.37 In addition, the progression of carcinoid heart disease can be retarded and prognosis improved.38
Hepatic arterial embolisation is based on the principle that hepatic metastases of NETs derive most of their blood supply from the hepatic artery. Embolisation with or without concomitant intra-arterial chemotherapy is effective in reducing tumour burden and controlling symptoms.39 The risk of provoking a “carcinoid crisis” is real, and pretreatment with an intravenous SSA is mandatory. Inadvertent embolisation of other organs (stomach, pancreas, and duodenum), with infarction as well as hepatic abscesses or local arterial injury, are all well documented adverse events. Embolisation with radioactive (90Yttrium) microbeads has been shown to have a significant effect in a limited number of patients, but serious adverse reactions have also been reported.40
The overall 5-year survival rate for patients with gastrointestinal NETs is about 58%, with little change over the past 30 years.7,29 However, in the subgroup of patients with well differentiated NETs with distant spread, the 5-year survival rate has improved from 15% to 52% over this time period. This improvement probably reflects the development of integrated management strategies, a multidisciplinary approach, and referral of patients with NETs to centres of expertise. It also coincides with the introduction of SSA therapy in the late 1980s. However, the paucity of large, well designed, comparative clinical trials, as well as the poor understanding of the natural history of these tumours, continues to be a problem and makes interpretation of such observational data difficult.
Despite the general assumption that gastrointestinal NETs are extremely rare, benign, slow-growing tumours, it is evident that they are far more common than previously thought and that, for some patients, the prognosis is poor. The non-specific nature of the symptoms and signs means they are often misinterpreted, and the diagnosis is usually delayed. This reflects a lack of physician training and public education. As a result, in the most common NETs, metastases are present in about 65% of cases at diagnosis.5 Although anatomical imaging is useful for accurate diagnosis, the lack of sensitive and specific plasma or genetic markers that could be used to screen for, predict or identify, early lesions or micrometastasis is a major impediment to precise diagnosis and optimal therapy.
1 Presenting complaints in patients with gastrointestinal neuroendocrine tumours and common misdiagnoses
 |
|
The non-specific nature of the symptoms and signs (inner circle) result in diagnostic error (outer circle) or delay in diagnosis.13 |
2 Utility of chromogranin A (CgA) as a marker of neuroendocrine tumours (NETs)
3 Clinical features and management of patients with gastrointestinal neuroendocrine tumours (NETs), by their primary organ site
Clinical features: NETs of the stomach are increasing in prevalence, probably as a result of the greater number of upper endoscopies performed, as well as a change in incidence.7,8 The aetiological importance of hypergastrinaemia, driven by proton-pump inhibitor use, is contentious.28
Clinical features: Patients with small-intestine NETs often present at a late stage, unless a mechanical event — obstruction, perforation, or bleeding — occurs. The prognosis is poor once tumours have spread beyond the intestine. Small-bowel NETs are commonly found in autopsy series, indicating that the true incidence is probably higher.9 Liver metastases with carcinoid syndrome are relatively common. Up to 50% of patients with small-bowel NETs have right-sided cardiac valvular disease,14 although this is now less common with the widespread use of somatostatin analogues and decreased serotonin levels. Mesenteric fibrosis may also cause mesenteric vessel occlusion and gastrointestinal ischaemia, perhaps from local secretion of serotonin or connective-tissue growth factor. About a fifth of patients with small-intestine NETs have synchronous or metachronous adenocarcinoma in the colon or elsewhere, and a quarter of small-intestine NETs are multicentric.
Clinical features: A relatively benign type of NET (7% of all gastrointestinal NETs) occurs in the appendix. It is usually identified serendipitously during appendicectomy or diagnostic gynaecological laparoscopy.29
Clinical features: NETs in the colon are common, especially in the caecum.7,8 The symptoms are similar to those of colon cancer — altered bowel habit, abdominal pain and bleeding. Concomitant carcinoid syndrome is extremely rare. The prognosis is relatively poor, with a 5-year survival rate of < 50%.7,8
Clinical features: In the United States, the rectum is involved in a quarter of patients with gastrointestinal NETs but, in Asia, this is the most common primary site.30 Half the NETs of the rectum are asymptomatic and diagnosed by colonoscopy for cancer screening or rectal bleeding. Endoscopic ultrasound is recommended for staging. These tumours are unpredictable, but generally have a low propensity to metastasise.
Management: NETs of the rectum are treated with endoscopic submucosal resection or surgery. The prognosis is excellent, with a 5-year survival rate of 85%.7,8
Clinical features: The incidence of pancreatic NETs is increasing.7,8 They are found incidentally by computed tomography imaging for unrelated abdominal complaints, or in response to symptoms of abdominal pain, diarrhoea, flushing and nausea from peptide release. Islet-cell carcinoma is the most common type. The overall prognosis is highly variable but, on average, is relatively poor (5-year survival rate, 27%).7,8
4 A summation of treatment efficacy in neuroendocrine tumours
5 Algorithm for the management of patients with suspected neuroendocrine tumours (NETs) of the small intestine
- Irvin M Modlin1
- Steven F Moss2
- Kjell Oberg3
- Robert Padbury4
- Rodney J Hicks5
- Bjorn I Gustafsson6
- Nicholas A Wright7
- Mark Kidd1
- 1 School of Medicine, Yale University, New Haven, Conn, USA.
- 2 Brown University, Rhode Island Hospital, Providence, RI, USA.
- 3 Uppsala University, Department of Medical Sciences, University Hospital, Uppsala, Sweden.
- 4 Flinders Medical Centre, Adelaide, SA.
- 5 University of Melbourne, Peter MacCallum Cancer Centre, Melbourne, VIC.
- 6 Norwegian University of Science and Technology, Department of Gastroenterology, St Olavs Hospital, Trondheim, Norway.
- 7 Barts and the London School of Medicine and Dentistry, London, UK.
Irvin Modlin has received speaker’s fees and travel support from Ipsen (2007), Novartis (2009) and Covidien (2009). Steven Moss has received travel support from Ipsen (2007), Tercica (2008) and Novartis (2009). Kjell Oberg has received travel assistance and speaker’s fees from Ipsen and Novartis (2009). Robert Padbury has received an honorarium from Novartis and travel support (2009). Rodney Hicks has spoken at Novartis- and Ipsen-sponsored meetings, but all honoraria were donated to his institution. He has received travel support from Novartis (2009). Bjorn Gustafsson has received speaker’s fees from Astra Zeneca (2009), and speaker’s fees and travel assistance from Ipsen (2009), as well as travel assistance from Novartis (2009). Mark Kidd has received travel support from Novartis (2009).
- 1. Modlin IM, Shapiro MD, Kidd M. Siegfried Oberndorfer: origins and perspectives of carcinoid tumors. Hum Pathol 2004; 35: 1440-1451.
- 2. Kloppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: the WHO classification. Ann N Y Acad Sci 2004; 1014: 13-27.
- 3. Ekeblad S, Skogseid B, Dunder K, et al. Prognostic factors and survival in 324 patients with pancreatic endocrine tumor treated at a single institution. Clin Cancer Res 2008; 14: 7798-7803.
- 4. Pape UF, Jann H, Muller-Nordhorn J, et al. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer 2008; 113: 256-265.
- 5. Modlin IM, Oberg K, Chung DC, et al. The current status of gastroenteropancreatic neuroendocrine tumours. Lancet Oncology 2008; 9: 61-72.
- 6. Plockinger U, Rindi G, Arnold R, et al. Guidelines for the diagnosis and treatment of neuroendocrine gastrointestinal tumours. A consensus statement on behalf of the European Neuroendocrine Tumour Society (ENETS). Neuroendocrinology 2004; 80: 394-424.
- 7. Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008; 26: 3063-3072.
- 8. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003; 97: 934-959.
- 9. Berge T, Linell F. Carcinoid tumours. Frequency in a defined population during a 12-year period. Acta Pathol Microbiol Scand A 1976; 84: 322-330.
- 10. Gustafsson BI, Kidd M, Modlin IM. Neuroendocrine tumors of the diffuse neuroendocrine system. Curr Opin Oncol 2008; 20: 1-12.
- 11. Kloppel G. Tumour biology and histopathology of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab 2007; 21: 15-31.
- 12. Klimstra DS, Modlin IM, Adsay NV, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am J Surg Pathol 2010; 34: 300-313.
- 13. Modlin IM, Kidd M, Latich I, et al. Current status of gastrointestinal carcinoids. Gastroenterology 2005; 128: 1717-1751.
- 14. Gustafsson BI, Hauso O, Drozdov I, et al. Carcinoid heart disease. Int J Cardiol 2008; 129: 318-324.
- 15. Stridsberg M, Oberg K, Li Q, et al. Measurement of chromogranin A, chromogranin B (secretogranin I), chromogranin C (secretogranin II) and pancreastatin in plasma and urine from patients with carcinoid tumours and endocrine pancreatic tumours. J Endocrinol 1995; 144: 49-59.
- 16. Sciarra A, Monti S, Gentile V, et al. Chromogranin A expression in familial versus sporadic prostate cancer. Urology 2005; 66: 1010-1014.
- 17. Syversen U, Ramstad H, Gamme K, et al. Clinical significance of elevated serum chromogranin A levels. Scand J Gastroenterol 2004; 39: 969-973.
- 18. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol 2005; 89: 151-160.
- 19. van der Horst-Schrivers AN, Post WJ, Kema IP, et al. Persistent low urinary excretion of 5-HIAA is a marker for favourable survival during follow-up in patients with disseminated midgut carcinoid tumours. Eur J Cancer 2007; 43: 2651-2657.
- 20. Bellutti M, Fry LC, Schmitt J, et al. Detection of neuroendocrine tumors of the small bowel by double balloon enteroscopy. Dig Dis Sci 2009; 54: 1050-1058.
- 21. Yamagishi H, Fukui H, Shirakawa K, et al. Early diagnosis and successful treatment of small-intestinal carcinoid tumor: useful combination of capsule endoscopy and double-balloon endoscopy. Endoscopy 2007; 39 Suppl 1: E243-E244.
- 22. Patel KK, Kim MK. Neuroendocrine tumors of the pancreas: endoscopic diagnosis. Curr Opin Gastroenterol 2008; 24: 638-642.
- 23. Hoeffel C, Job L, Ladam-Marcus V, et al. Detection of hepatic metastases from carcinoid tumor: prospective evaluation of contrast-enhanced ultrasonography. Dig Dis Sci 2009; 54: 2040-2046.
- 24. Pantongrag-Brown L, Buetow PC, Carr NJ, et al. Calcification and fibrosis in mesenteric carcinoid tumor: CT findings and pathologic correlation. AJR Am J Roentgenol 1995; 164: 387-391.
- 25. Janson ET. Somatostatin analogs in the treatment of neuroendocrine gastroenteropancreatic and intrathoracic tumors. J Endocrinol Invest 2005; 28: 137-140.
- 26. Marko J, Lamba R, Miller F, et al. OctreoScan positive Crohn’s disease mimicking an ileal carcinoid tumor. J Clin Gastroenterol 2008; 42: 66-68.
- 27. Hofmann M, Maecke H, Borner R, et al. Biokinetics and imaging with the somatostatin receptor PET radioligand (68)Ga-DOTATOC: preliminary data. Eur J Nucl Med 2001; 28: 1751-1757.
- 28. Modlin IM, Lye KD, Kidd M. A 50-year analysis of 562 gastric carcinoids: small tumor or larger problem? Am J Gastroenterol 2004; 99: 23-32.
- 29. Modlin IM, Champaneria MC, Chan AK, Kidd M. A three-decade analysis of 3,911 small intestinal neuroendocrine tumors: the rapid pace of no progress. Am J Gastroenterol 2007; 102: 1464-1473.
- 30. Li AF, Hsu CY, Li A, et al. A 35-year retrospective study of carcinoid tumors in Taiwan: differences in distribution with a high probability of associated second primary malignancies. Cancer 2008; 112: 274-283.
- 31. Modlin IM, Moss SF, Chung DC, et al. Priorities for improving the management of gastroenteropancreatic neuroendocrine tumors. J Natl Cancer Inst 2008; 100: 1282-1289.
- 32. Kerstrom G, Hellman P, Hessman O. Midgut carcinoid tumours: surgical treatment and prognosis. Best Pract Res Clin Gastroenterol 2005; 19: 717-728.
- 33. Dierdorf SF. Carcinoid tumor and carcinoid syndrome. Curr Opin Anaesthesiol 2003; 16: 343-347.
- 34. Schmid HA. Pasireotide (SOM230): development, mechanism of action and potential applications. Mol Cell Endocrinol 2008; 286: 69-74.
- 35. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009; 27: 4656-4663.
- 36. Van Essen M, Krenning EP, De Jong M, et al. Peptide receptor radionuclide therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol 2007; 46: 723-734.
- 37. Landry CS, Scoggins CR, McMasters KM, Martin RC, 2nd. Management of hepatic metastasis of gastrointestinal carcinoid tumors. J Surg Oncol 2008; 97: 253-258.
- 38. Moller JE, Pellikka PA, Bernheim AM, et al. Prognosis of carcinoid heart disease: analysis of 200 cases over two decades. Circulation 2005; 112: 3320-3327.
- 39. Liapi E, Geschwind JF, Vossen JA, et al. Functional MRI evaluation of tumor response in patients with neuroendocrine hepatic metastasis treated with transcatheter arterial chemoembolization. AJR Am J Roentgenol 2008; 190: 67-73.
- 40. Murthy R, Nunez R, Szklaruk J, et al. Yttrium-90 microsphere therapy for hepatic malignancy: devices, indications, technical considerations, and potential complications. Radiographics 2005; 25 Suppl 1: S41-S55.
Abstract
Neuroendocrine tumours (NETs) are increasing in both incidence and prevalence and, as a group, are more prevalent than either gastric, pancreatic, oesophageal or hepatobiliary adenocarcinomas, or any two of these cancers combined.
Clinical awareness of the protean and intermittent symptoms of NETs (eg, sweating, flushing, diarrhoea, and bronchospasm) is critical for timely diagnosis; however, the classical carcinoid syndrome is relatively uncommon.
The most useful diagnostic test for gastrointestinal NETs is measurement of plasma chromogranin A (CgA) levels. Disease extent is assessed by both anatomical imaging, and nuclear imaging with radiolabelled somatostatin analogues.
Pathological evaluation comprises tumour–node–metastasis classification, a minimum pathological dataset, CgA and synaptophysin immunostaining, as well as mitotic count or Ki-67 index (a marker of cell proliferation) to define grading.
Resection of the primary lesion and as much metastatic disease as possible increases the efficacy of medical therapy. Other management strategies include hepatic embolisation and peptide receptor radionuclide therapy.
Patients with tumours expressing somatostatin receptors should be treated with somatostatin analogues. Depending on the tumour grade, other effective agents include cytotoxics, tyrosine kinase inhibitors, and antiangiogenics.
The overarching requirement for best management of patients with NETs is to ensure that they have ready access to experienced multidisciplinary clinician groups located within centres of appropriate subspecialty expertise.