





Volume 190 - Issue 6
Clinical, electrophysiological and genetic features of a large Australian family with paramyotonia congenita
Authors: Sharavanan Parasivam, Malgorzata Krupa, Mark Slee and Dominic E Thyagarajan
Med J Aust 2009; 190 (6): 334-336. || doi: 10.5694/j.1326-5377.2009.tb02428.x
Published online: 16 March 2009
Published online: 16 March 2009
A 32-year-old woman with a 4-year history of multiple sclerosis presented with persistent clawing of the right hand. History revealed that she and five family members had lifelong symptoms of paradoxical myotonia (impaired relaxation of muscles following muscle contraction), exacerbated by cold. The family was diagnosed with paramyotonia congenita, based on neurophysiological and genetic studies. To our knowledge, this is the first report of an Australian family with paramyotonia congenita.
Clinical record
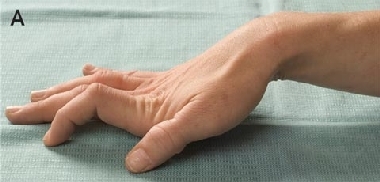
A 32-year-old woman of European ancestry was referred to a movement disorder clinic for evaluation of mild persistent clawing of the right hand (Figure, A), which had developed over the past year. She had no weakness of the hand or other neurological signs.
The patient had been diagnosed with relapsing remitting multiple sclerosis 4 years earlier, based on clinical features (typical exacerbations), characteristic white matter changes on magnetic resonance imaging scans, and oligoclonal bands restricted to the cerebrospinal fluid. Her condition was managed in a multiple sclerosis clinic. Before her referral, she was in good health between exacerbations of multiple sclerosis.
Since early childhood, the patient had experienced persistent cramping of her hands after their use, particularly in cold conditions. She had difficulty releasing tightly gripped objects, and cramping usually worsened, rather than improved, with ongoing exertion, such as when using clippers to groom a dog (ie, there was no “warm-up” phenomenon). She found it difficult to talk after ingesting cold food or drink, and difficult to open her eyes after jumping into a cold pool. In extremely cold conditions, she experienced widespread cramping of her muscles with clawing of her hands, and a tendency for her toes to curl inwards. These symptoms would typically improve over several hours as her body warmed. As the cramping improved, there was notable weakness of affected muscles. A diagnosis of paramyotonia congenita was suspected after referral to the clinic.
On questioning, the patient reported that other members of her family had similar symptoms. Five were interviewed; their symptoms began in early childhood, and included muscle cramping induced by exposure to cold or exertion (both in most cases). Face and hand muscles were predominantly affected. These family members also described generalised cramping and weakness in response to severe cold, but none had a permanent deformity similar to the patient’s clawing of the right hand.
Physical examination of the patient and five affected family members revealed that all had paradoxical myotonia of the orbicularis oculi muscles and the hands, and percussion myotonia over the thenar eminence. None had weakness or muscle hypertrophy. Owing to the unusual coincidence of white matter disease and suspected paramyotonia congenita in the patient, magnetic resonance imaging of the proband’s affected brother (Figure, B; V-4) was performed. A small area of gliosis in the left caudate nucleus was identified but no white matter abnormalities were present.
A family pedigree was constructed (Figure, B). This revealed that the paradoxical myotonia was inherited in an autosomal dominant pattern, spanning at least six generations.
Further investigations of the patient revealed a slightly elevated creatine kinase level, myotonic discharges on electromyography and evidence of cold paralysis on nerve conduction studies. DNA sequencing revealed that the patient had a sodium channel gene mutation (Box). The five affected family members were subsequently identified as being heterozygous for the same mutation, and an unaffected family member was shown to lack this mutation (Box). Paramyotonia congenita was diagnosed, and acetazolamide therapy was begun, which led to moderate amelioration of symptoms. Her multiple sclerosis remained well controlled on glatiramer acetate.
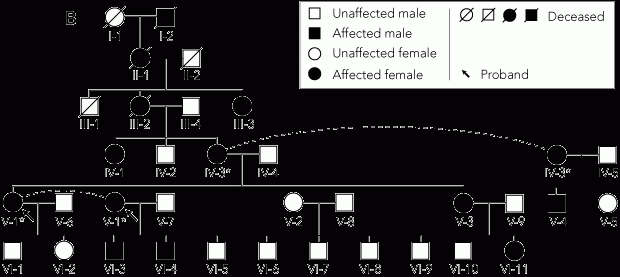 | |||||||||||||||
* Individuals IV-3 and V-1 each appear twice in the pedigree, as indicated by the dashed lines. | |||||||||||||||
To our knowledge, this is the first report of an Australian family with paramyotonia congenita. Paramyotonia congenita is a rare autosomal dominant condition characterised by paradoxical myotonia — impaired relaxation of muscles following muscle contraction, which worsens with repetitive muscle activity.1 It is distinct from other forms of myotonia, which typically improve with repetitive activity (the warm-up phenomenon).4 Paramyotonia is typically exacerbated by exposure to cold, and can be associated with cold-induced paralysis.1 Mutations in the skeletal muscle voltage-gated sodium channel gene SCN4A cause paramyotonia congenita,5 and can also cause hyperkalaemic periodic paralysis, potassium-aggravated myotonia, and a small proportion of hypokalaemic periodic paralysis cases.6 Patients carrying an SCN4A mutation may have manifestations of more than one of these allelic disorders.
Our patient’s neurophysiology findings and the clinical features of her affected family members are typical for paramyotonia congenita associated with cold paralysis, and similar to those described in the largest reported case series for this condition.7 In this case series, age of onset was typically during early childhood, and clinical myotonia was evident in 100% of the 56 patients studied. Cold was identified as a precipitant in 91% of patients, and exercise was identified as a precipitant in 46%. Electromyographic evidence for myotonia was seen in 100% of the patients, and 92% had a reduction in compound muscle action potential in response to cold (objective cold paralysis). Forty-nine of the patients (88%) had a mutation of the SCN4A gene. The electromyographic changes evident with cooling in our patient were typical for paramyotonia congenita — cooling initially resulted in abolition of the myotonic discharges, and electrical silence was recorded at lower temperatures.1
The T1313M mutation was identified in our patient and the five affected family members, but not an unaffected family member. This is one of the more common mutations that causes paramyotonia congenita,4,8,9 and has been reported to exclusively cause paramyotonia with the cold-paralysis phenotype.9-13 The hands and face are predominantly affected in patients who carry the T1313M mutation.9-13 A report of French families with paramyotonia congenita noted significant clinical variability in patients carrying the T1313M mutation — in both severity of myotonia and its permanence — and myotonia permanens was evident in six of eight of these patients.9 In our patient, the clawing of the right hand is also likely to reflect myotonia permanens, rather than an interaction between the paramyotonia and multiple sclerosis.
The T1313M mutation has been predominantly described in families with French ancestry, and to a lesser extent in families of English and Japanese background. 5,9,11 One de-novo mutation has been identified in the literature, which occurred in a Japanese man.12 All individuals from the six generations of the family we studied were born in Australia and lived in Australia, but the ancestral origins of the family are unknown.
Present knowledge of SCN4A channel physiology provides insight into the clinical manifestations seen in our patient and the affected members of her family. Voltage-gated sodium channels are heteromultimeric, integral membrane proteins; they are composed of a single large pore-forming α subunit, and 1 or 2 smaller β units.6 There are nine subtypes of α subunit, one of which is expressed in skeletal muscle — SCN4A.6 The α subunit is composed of four structurally homologous domains (D1–D4), with six membrane spanning segments (S1–S6) within each domain.6 The sodium channel is important for generating and propagating action potentials and switches through three functional states: activation (the open-channel state), inactivation, and recovery from inactivation.6
The consequences of the T1313M mutation have been studied by patch-clamp studies using various cell lines. 10,14-17 Overall, the mutation has been shown to slow the rate of channel inactivation, diminish the voltage dependence of inactivation, and increase the rate of recovery from inactivation. 10,14-17 These effects result in increased sodium conductance and prolongation of action potentials, which causes persistent activation of potassium channels, leading to relatively high levels of extracellular potassium.6,10,14-17 Elevated extracellular potassium levels increase the likelihood of afterdepolarisations, which may lead to action potentials on adjacent surface membranes.6 This can result in ongoing muscle contraction and slowed relaxation — the features of paramyotonia.6
Threonine-1313 is a highly conserved residue in the D3–D4 linker; it is important for channel inactivation, and thought to occlude the cytoplasmic portion of the channel.16 The change from the polar hydrophilic threonine to the larger non-polar methionine is thought to impair occlusion of the channel, and therefore impair inactivation.13,16 Although the effects of the mutation have been shown to be potentiated at lower temperatures, as might be expected, no study has shown an increased sensitivity to temperature compared with normal cells.15-17
We have described the clinical, neurophysiological and genetic features of a large Australian family with paramyotonia congenita. The diagnosis of myotonia may be difficult and cause confusion, and the sodium channel disorder described here should be considered in patients with the symptoms described — especially as the symptoms respond to treatments such as acetazolamide and mexiletine, and as there are implications for genetic counselling and prognosis. Clues to the diagnosis include paradoxical myotonia — myotonia that worsens with ongoing exertion — and significant worsening of symptoms in cold conditions.
Investigations
Biochemistry and neurophysiology
The patient’s serum potassium level was 4.2 mmol/L (reference range [RR], 3.2–4.3 mmol/L) and her creatine kinase level was slightly elevated at 191 U/L (RR, < 150 U/L). Routine nerve conduction study results were normal. Electromyography of the left abductor pollicis brevis and flexor digitorum superficialis revealed electrical myotonia and dense fibrillations. Cooling to below 28°C abolished the myotonic discharges and cooling below 22°C resulted in abolition of all spontaneous activity. The cooling test described by Streib1 was also performed. Before cooling, the amplitude of the left median compound muscle action potential recording over the abductor pollicis brevis muscle was 16.7 mV. The arm was then cooled to below 20°C by immersion in an ice bath and then rewarmed to 32°C using heat packs. The compound muscle action potential after rewarming was 4.5 mV (a 73% decrease).
Venous blood from the patient was sent to a commercial laboratory (PathWest, Royal Perth Hospital, Perth) for sequencing of the SCN4A gene. A heterozygous C-to-T nucleotide substitution at position 3938 in exon 22 was identified, resulting in a threonine-to-methionine amino acid change at codon 1313 (T1313M).
Venous blood was then obtained from five family members who had symptoms that were similar to those of the patient (Figure, B; IV-1, IV-3, V-3, V-4, VI-4), as well as an unaffected family member (the patient’s half-sister), and mutational analysis was carried out. DNA was extracted by a proteinase K digestion method2 and amplified using the polymerase chain reaction (PCR) (forward primer sequence, 5'-TGGAGGCAGGAAGGGGAACT-3'; reverse primer sequence, 5'-GGCAGCACACACAGGACAGG-3'). The cycling conditions were: 3 min at 94°C × 1; and 30 s at 94°C, 40 s at 57°C, 1 min at 72°C × 30. The PCR reaction mixture contained 100 ng DNA, 0.5 μM of each primer, 80 μM of each deoxynucleoside triphosphate, 20 μM Tris-HCl (pH, 8.5), 50 μM KCI, 1.5 mM MgCI and 0.5 U Taq Ti DNA polymerase (Fisher Biotech, Perth, Australia).
Amplified products were separated on 3% agarose gels. The separated fragments were diluted to 10 ng/μL per 100 base pairs and then sequenced by the dideoxy termination method,3 using BigDye Terminator v3.1 chemistry (Applied Biosystems, Foster City, Calif, USA) and the 3100 Genetic Analyzer (Applied Biosystems).
Sequencing results showed that the five affected family members were heterozygous for the T1313M mutation and that the unaffected family member did not carry the mutation.
Competing interests
None identified.
Acknowledgements
We thank the patient and her family for consenting to publication of this case report. Publication was also approved by the Flinders Clinical Research Ethics Committee.
References
- Streib EW. AAEE minimonograph #27: differential diagnosis of myotonic syndromes. Muscle Nerve 1987; 10: 603-615. 0_i1092008
- Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988; 38: 1339-1346. 0_i1092010
- Sanger F, Nicklen S, Coulson A. DNA sequencing with chain terminating inhibitors. Proc Natl Acad Sci U S A 1977; 74: 5463-5467. 0_i1092012
- Jurkat-Rott K, Lehmann-Horn F. Muscle channelopathies and critical points in functional and genetic studies. J Clin Invest 2005; 115: 2000-2009. 0_i1092014
- McClatchey A, Van den Bergh P, Pericak-Vance M, et al. Temperature-sensitive mutations in the III–IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita. Cell 1992; 68: 769-774. 0_i1092016
- George L. Inherited disorders of voltage gated sodium channels. J Clin Invest 2005; 115: 1990-1999. 0_i1092018
- Miller M, Dias da Silva M, Miller H, et al. Correlating phenotype and genotype in the periodic paralyses. Neurology 2004; 63: 1647-1655. 0_i1092020
- Ptacek L. The familial periodic paralyses and nondystrophic myotonias. Am J Med 1998; 105: 58-70. 0_i1092022
- Plassart E, Reboul J, Rime C, et al. Mutations in the muscle sodium channel gene (SCN4A) in 13 French families with hyperkalaemic periodic paralysis and paramyotonia congenita: phenotype to genotype correlations and demonstration of the predominance of two mutations. Eur J Hum Genet 1994; 2: 110-124. 0_i1092024
- Tahmoush A, Schaller K, Zhang P, et al. Muscle sodium channel inactivation defect in paramyotonia congenita with the thr1313met mutation. Neuromusc Disord 1994; 4: 447-454. 0_i1092026
- Yamada T, Ochi H, Hara H, et al. A skeletal muscle sodium channel mutation in a Japanese family with paramyotonia congenita. J Neurol Sci 1995; 133: 192-193. 0_i1092028
- Fukudome T, Izumoto H, Goto H, et al. Paramyotonia congenita due to a de novo mutation: a case report. Muscle Nerve 2003; 28: 232-235. 0_i1092030
- Bouhours M, Sternberg D, Davoine C, et al. Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans. J Physiol 2003; 554: 635-647. 0_i1092032
- Boulos P, Heiman-Patterson T, Alexander G, Tahmoush A. Patch clamp studies of the thr1313met mutant sodium channel causing paramyotonia congenita. Muscle Nerve 2000; 23: 1736-1747. 0_i1092034
- Dice M, Abbruzzese J, Wheeler J, et al. Temperature-sensitive mutants in paramyotonia congenita mutants R1448C and T1313M. Muscle Nerve 2004; 30: 277-288. 0_i1092036
- Hayward L, Brown R, Cannon S. Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. J Gen Physiol 1996; 107: 559-576. 0_i1092038
- Yang N, Ji S, Zhou M, et al. Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc Natl Acad Sci U S A 1994; 91: 12785-12789. 0_i1092042