





Volume 190 - Issue 4 Supplement
Synapse formation and regression in the cortex during adolescence and in schizophrenia
Author: Maxwell R Bennett
Med J Aust 2009; 190 (4 Suppl): S14. || doi: 10.5694/j.1326-5377.2009.tb02368.x
Published online: 16 February 2009
Published online: 16 February 2009
Abstract
During adolescence, about 30% of the synapses formed during childhood in the dorsolateral prefrontal cortex (DLPC) are lost; in patients with schizophrenia, the synapse loss is about 60%.
Studies of synapse loss in the neuromuscular junction have provided insights into the molecular basis of synapse formation and regression, thereby providing a paradigm for investigations of synapse loss in the DLPC.
Research into some of these molecules in the DLPC has shown that they are crucial to synapse formation and regression.
Further research in this field could examine when synapse loss in the DLPC of patients with schizophrenia occurs, and further elucidate how these molecules are involved in the development of schizophrenia.
Changes in synapse density in the supragranular levels (layer III) of the human dorsolateral prefrontal cortex (DLPC) during development were first reported in 1979.1 The study found that synapse density reaches a peak before adolescence, then decreases by at least 30% during the adolescent years, and remains constant thereafter until later in life (> 70 years).1
More recently, studies using antibodies to synaptic vesicle-associated proteins have confirmed this synapse loss during adolescence. They have also shown that the number of synapses in the DLPC is greatly reduced in patients with schizophrenia, beyond the 30% loss that normally occurs during adolescence; no such enhanced loss occurs in the visual cortex of such patients.2,3 The mechanisms underpinning this synapse loss is of great interest, particularly as decreasing connectivity below a certain value in the neural networks of the brain is expected to lead to rapid loss of function.4
A key question for schizophrenia research is: does the smaller number of synapses observed in the DLPC of patients with schizophrenia arise as a consequence of a failure of synapses to form before adolescence, or does it occur as a consequence of an excess pruning of synapses during adolescence?5 A study of patients at high risk of developing schizophrenia found that adolescents in the prodromal phase lost more grey matter in the DLPC compared with adolescents at baseline levels of risk,6 suggesting that an excess loss of synapses does occur during adolescence. Present knowledge supports the hypothesis that there is excessive regression of synapses in the DLPC of patients with schizophrenia, especially of layer III pyramidal neurones receiving corticocortical and thalamic-cortical projections; whether this occurs before, during or after adolescence is not clear.
Here, I examine what molecular events might occur at synapses in the DLPC that are involved in their formation and regression, using our knowledge of the neuromuscular synapse as a paradigm.
Molecular determinants of synapse formation and regression
At the neuromuscular junction
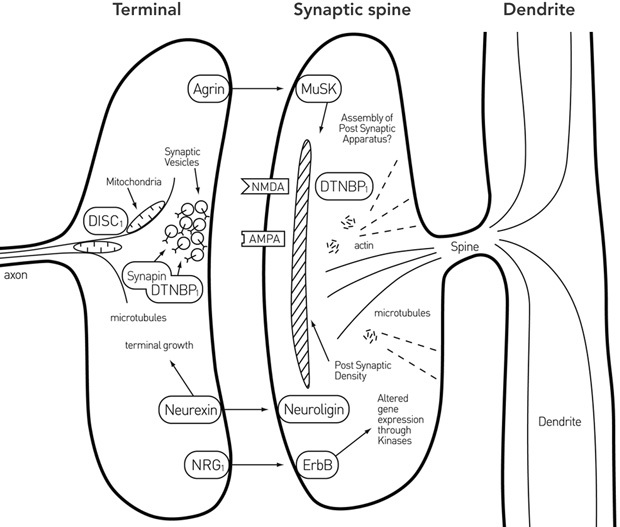
Over the past 30 years, a number of molecules have been identified that are central to the process of synapse formation and regression at the neuromuscular junction (Box 1). These include muscle-specific kinase (MuSK), agrin, the dystroglycan–utrophin–dystrophin complex (of which dysbindin 1 is a component) and neuregulin (NRG), together with other molecules of interest such as DISC1 (disrupted in schizophrenia 1) and neurexin (Box 2). I examine the role at the neuromuscular junction of each of these in turn and consider their normal function at synapses in the DLPC, together with what might have gone wrong with such functions in schizophrenia. An excessive loss of synapses as a result of loss of function of these molecules is known, on theoretical grounds, to lead to a failure of neural network function, and may then contribute to what has gone awry in schizophrenia.
In central synapses
MuSK, the principal molecule involved in neuromuscular synaptic development, has recently been found in neurones throughout the cortex. MuSK and agrin are found in about 30% of all excitatory synapses on neurones in the brain.13
It is therefore of great interest that, in agrin-deficient mice, dysregulation of components of the MuSK pathway occurs and there is about a 30% loss of synapses.13 Unfortunately, it is not known when this loss occurs in relation to adolescence. However, as conditional inactivation of MuSK in adult animals leads to loss of neuromuscular synapses, it would be anticipated that in agrin-deficient mice, up to 30% of excitatory cortical synapses would not form at all. If such results apply to humans, it would be expected that during childhood about 30% of excitatory synapses would fail to form, which is the percentage missing in the DLPC of adults with schizophrenia.
DTNBP1 (dystrobrevin-binding protein 1; dysbindin 1) is found in the cortex in association with microtubules and postsynaptic densities of spines at certain synapses, as well as in glutamatergic nerve terminals in association with the protein synaptin, which primes synaptic vesicles for exocytosis. Overexpression of DTNBP1 leads to enhanced glutamate release at synapses, whereas underexpression does the opposite,14 possibly reflecting the influence of DTNBP1 on synaptin’s control of vesicle priming.
NRG1 is localised in cortical neurones and their synaptic terminals on spines, like it is in motor neurones and their terminals at synapses on muscle cells. Preventing NRG1–ErbB4 signalling via ErbB4 RNA interference destabilises AMPA receptors and leads to loss of synaptic NMDA currents and decrease in spine size. The eventual loss of synaptic NMDA currents from mature neurones is about 50%.15 It is clear that NRG1–ErbB4 dysfunction during development will lead to a significant loss of excitatory synapse formation in the DLPC during childhood.
DISC1 immunoreactivity is found at synapses of dendritic spines in human prefrontal and parietal cortex. DISC1 is localised to mitochondria in cortical neurones, and its complex with FEZ1 (fasciculation and elongation protein zeta 1) is necessary for transport of these mitochondria from soma to axon and dendrites. Without this transport, the neurones do not form neurites.16 The implications of this for synapse formation are likely to be considerable, but are still unclear.
Neurexin is found in nerve terminals with α-neurexin and binds to postsynaptic neuroligin to ensure synaptic terminal growth. There is a 20% loss of neuropile in α-neurexin knockout mice, which is primarily due to loss of inhibitory type II synapses.17 A lack of neurexin would thus be expected to lead to significant failure of inhibitory synapse formation in the DLPC during childhood.
Loss of synapse formation and regression molecules in the DLPC of patients with schizophrenia
No study has yet reported changes in the expression of MuSK or agrin in the cortices of patients with schizophrenia. Given the importance of these molecules, it is clearly of interest to determine if such changes occur in these individuals.
In patients with schizophrenia, DTNBP1 messenger RNA (mRNA) is significantly decreased in supragranular layers II and III and infragranular layers V and VI, but not granular layer IV; no differences in mRNA for a variety of other proteins (spinophilin, synaptophysin, cyclophilin) have been detected in any DLPC layer.18 Loss of DTNBP1 results in a considerable decrease in glutamate release from synapses.14 It is therefore likely that DTNBP1 reduction in patients with schizophrenia leads to a loss in the efficacy of excitatory synaptic transmission in at least the DLPC.
NRG1a is significantly decreased in the DLPC grey matter of patients with schizophrenia, although NRG1b may be increased.19 A loss of the NRG1–ErbB4 interaction could lead to failure of excitatory synapse formation of up to 50%.15
The Scottish study that led to the discovery of DISC1 involved a translocation that removes interaction between NDEL1 (nuclear disruption element-like 1; also known as NUDEL) and DISC1.20 Indeed, single nucleotide polymorphisms in the DISC1 gene associated with schizophrenia give rise to reduced grey matter volume in the prefrontal cortex,21 possibly due to the loss of synaptic dendritic spines, and of entire basilar and apical dendrites.
Conclusion
When neuromuscular synapses form during development, a phalanx of molecules is orchestrated in the process, including MuSK, agrin, NRG and ErbB4. It is argued that competition between nerve terminals during the subsequent period of synapse regression involves competition for these molecules. It is highly likely that these molecules are also important in synapse formation in the DLPC during childhood, and perhaps in synapse regression during adolescence. Further, we have seen that if these molecules are not expressed, about 30% of synapses fail to form at all in the cortex. This being the case, it is expected that a failure of many synapses to form during childhood, rather than an excess loss of synapses during adolescence, might characterise schizophrenia.
Different clinical stages have been identified during the prodromal period for schizophrenia.22 Are these related to synapse loss in the DLPC? It is difficult to see how this could be the case if the relatively small number of synapses in the DLPC arises from a failure of synapses to form during childhood. On the other hand, such a relationship might arise as a consequence of the vulnerability of an already small number of synapses to further regress when such prodromal individuals are subjected to psychological stress, or stress due to infection. This could lead to a loss of synapses below the level at which DLPC networks can function.
This article is an attempt to focus attention on the possibility that our knowledge of synapse formation and regression at the neuromuscular junction, and its molecular requirements, can be used to guide us in understanding these phenomena in the DLPC. The major questions posed here are open to experimental determination:
Over what period are the synapse numbers in the DLPC of patients with schizophrenia lower than normal?
What is the relative importance of MuSK–agrin, neuregulin–ErbB4, neurexin–neuroligin and DISC1 in synapse formation and regression in the DLPC?
To what extent have these molecules gone awry in schizophrenia?
1 Key molecules in neuromuscular synapse formation and regression
Muscle-specific kinase (MuSK) in the postsynaptic membrane is activated by the proteoglycan agrin, which is released from motor–nerve terminals to generate synaptic-specific transcription. Inactivation of MuSK in embryonic mice leads to the failure of neuromuscular synapse formation. Conditional inactivation of MuSK in adult mice leads to loss of acetylcholine receptors, disassembly of the postsynaptic apparatus and regression of the synaptic terminal.7
Agrin-lacking mice fail to cluster acetylcholine receptors and form mature neuromuscular synapses, so they die at birth.8
Neuregulin 1 (NRG1), when released from the motor–nerve terminal, binds to its postsynaptic receptor ErbB4, leading to additional control of the number of acetylcholine receptors. The NRG1–ErbB4 interaction is required for differentiation of the postsynaptic apparatus.9
DISC1 (disrupted in schizophrenia 1) is found in peripheral neurones, both sensory and sympathetic-like, that form autonomic neuromuscular junctions.10 DISC1 interacts with FEZ1 (fasciculation and elongation protein zeta 1) and NDEL1 (nuclear disruption element-like 1; also known as NUDEL).11 This interaction involves DISC1 acting as a cargo receptor for regulating the localisation of the NDEL1–LIS1 (lissencephaly 1) complex into axons and growth cones necessary for axon elongation.
Neurexin is a presynaptic receptor at the neuromuscular junction. It binds α-latrotoxin (a major component of black widow spider venom) to produce a massive transmitter release. Although not directly involved in neuromuscular synapse formation, it is involved in the formation of central synapses.
2 Spatial distribution of molecules implicated in synapse formation and regression
It is proposed that the principal among these are MuSK and its nerve terminal released ligand agrin (MuSK–agrin), first discovered at the neuromuscular junction, as were nerve terminal released NRG and its receptor ErbB (NRG1–ErbB) and DTNBP1, but not DISC1, which is involved in microtubule formation (among other things).
DISC1 = disrupted in schizophrenia 1. DTNBP1 = dystrobrevin-binding protein 1 (dysbindin 1). MuSK = muscle-specific kinase. NRG1 = neuregulin 1. Reproduced with permission from Bennett MR. Aust N Z J Psychiatry 2008; 42: 662-677.12
Competing interests
None identified.
References
- Huttenlocher PR. Synaptic density in human frontal cortex — development changes and effects of aging. Brain Res 1979; 163: 195-205. 0_i1091938
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000; 57: 65-73. 0_i1091940
- Glantz LA, Gilmore JH, Hamer RM, et al. Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience 2007; 149: 582-591. 0_i1091942
- McGlashan TH, Hoffman RE. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiat 2000; 57: 637-648. 0_i1091944
- Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiat Res 1982; 17: 319-334. 0_i1091946
- Pantelis C, Velakoulis D, Wood SJ, et al. Neuroimaging and emerging psychotic disorders: the Melbourne ultra-high risk studies. Int Rev Psychiatry 2007; 19: 371-381. 0_i1091948
- Hesser BA, Henschel O, Witzemann V. Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol Cell Neurosci 2006; 31: 470-480. 0_i1091950
- Misgeld T, Kummer TT, Lichtman JW, Sanes JR. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proc Natl Acad Sci U S A 2005; 102: 11088-11093. 0_i1091952
- Kim N, Burden SJ. MuSK controls where axons grow and form synapses. Nat Neurosci 2008; 11: 19-27. 0_i1091954
- Grabham PW, Seale GE, Bennecib M, et al. Cytoplasmic dynein and LIS1 are required for microtubule advance during growth cone remodeling and fast axonal outgrowth. J Neurosci 2007; 27: 5823-5834. 0_i1091956
- Mackie S, Millar JK, Porteous DJ. Role of DISC1 in neural development and schizophrenia. Curr Opin Neurobiol 2007; 17: 95-102. 0_i1091958
- Bennett MR. Dual constraints on synapse formation and regression in schizophrenia: neuregulin, neuroligin, dysbindin, DISC1, MuSK and agrin. Aust N Z J Psychiatry 2008; 42: 662-677. 0_i1091960
- Ksiazek I, Burkhardt C, Lin S, et al. Synapse loss in cortex of agrin-deficient mice after genetic rescue of perinatal death. J Neurosci 2007; 27: 7183-7195. 0_i1091962
- Numakawa T, Yagasaki Y, Ishimoto T, et al. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet 2004; 13: 2699-2708. 0_i1091964
- Lin B, Woo RS, Mei L, Malinow R. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron 2007; 54: 583-597. 0_i1091966
- Ikuta J, Maturana A, Fujita T, et al. Fasciculation and elongation protein zeta-1 (FEZ1) participates in the polarization of hippocampal neuron by controlling the mitochondrial motility. Biochem Biophys Res Commun 2007; 353: 127-132. 0_i1091968
- Dudanova I, Taabuchi K, Rohlmann A, et al. Deletion of alpha-neurexins does not cause a major impairment of axonal pathfinding or synapse formation. J Comp Neurol 2007; 502: 261-274. 0_i1091970
- Weickert CS, Straub RE, McClintock BW, et al. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry 2004; 61: 544-555. 0_i1091972
- Chong VZ, Thompson M, Beltaifa S, et al. Elevated neuregulin-1 and ErbB4 protein in the prefrontal cortex of schizophrenic patients. Schizophr Res 2008; 100: 270-280. 0_i1091974
- Brandon NJ, Handford EJ, Schurov I, et al. Disrupted In Schizophrenia 1 and Nudel form a neurodevelopmentally regulated protein complex: implications for schizophrenia and other major neurological disorders. Mol Cell Neurosci 2004; 25: 42-55. 0_i1091976
- Mitelman SA, Brickman AM, Shihabuddin L, et al. A comprehensive assessment of gray and white matter volumes and their relationship to outcome and severity in schizophrenia. Neuroimage 2007; 37: 449-462. 0_i1091978
- McGorry PD, Purcell R, Hickie IB, et al. Clinical staging: a heuristic model for psychiatry and youth mental health. Med J Aust 2007; 187 (7 Suppl): S40-S42. 0_i1091980