





Volume 190 - Issue 11
Hysterectomy in a phenotypic male with advanced gonadal malignancy and intersex
Authors: Jim L Parker, Deborah L Ekman and Lawrence J Hayden
Med J Aust 2009; 190 (11): 644-646. || doi: 10.5694/j.1326-5377.2009.tb02595.x
Published online: 1 June 2009
Published online: 1 June 2009
Disorders of sex development (DSD), previously termed intersex, are uncommon, and are usually, but not always, diagnosed at birth. Issues of gender assignment, psychosexual development and the potential for malignant change in a dysgenetic gonad need to be considered. Here, we report a rare presentation of advanced malignancy in an abdominal gonad associated with the formation of a uterus in an adult male with a previously undiagnosed DSD.
Clinical record
A 59-year-old man presented with haematuria and gynaecomastia, and was subsequently found to have a large pelvic mass. Examination of his left breast identified a 3–4 cm mobile mass. Ultrasonography of the breast confirmed a mass, but could not distinguish between glandular tissue and a breast tumour. Computed tomography (CT) of the abdomen and pelvis showed a 10 cm complex pelvic mass that was thought to extend to the seminal vesicles. A core biopsy showed mixed cytological and immunohistochemical features of a possible granulosa cell tumour, but the precise diagnosis was limited by the small amount of material present for examination. A provisional diagnosis of gonadal malignancy in a dysgenetic gonad was considered.
A bone scan was negative and a thoracic CT scan showed three 5–7 mm lesions in the lung fields, suggestive of possible metastases. His prostate-specific antigen (PSA) level was very low (0.01 ng/mL), and tests for testicular tumour markers were negative (alpha-fetoprotein, 3 ng/mL; human chorionic gonadotropin, < 5 U/L). Other biochemical tests showed the following levels: serum oestradiol, 372 pmol/L (male reference range [RR], < 160 pmol/L); testosterone, 1.1 nmol/L (RR, 8–38 nmol/L); follicle-stimulating hormone, < 0.1 U/L (male RR, 1.0–9.2 U/L); luteinising hormone, 2.1 U/L (male RR, 2–8 U/L); prolactin, 385 mU/L (RR, 0–500 mU/L).
The patient had a history of penoscrotal hypospadias, cryptorchidism and right inguinal hernia that were surgically repaired in infancy. The hypospadias was corrected at 20 months of age. He had a right inguinal hernia repair at 3 years of age and, at that time, was found to have an undescended right testis that was treated by orchidopexy. The patient’s parents were told that the left testis was removed during this procedure.
He was married with no children, and we do not know whether the issue of fertility had ever been questioned. He was noted to have minor obstructive urinary symptoms at 47 years of age that were thought to be caused by a urethral stricture secondary to his previous hypospadias repair.
He re-presented 8 years later with a bloody urethral discharge that settled after treatment with antibiotics. On rectal examination at age 55, he had a small prostate and his PSA level was low (0.02 ng/mL). At that time, an intravenous pyelogram showed normal kidneys, ureters and bladder. At cystoscopy, he was found to have a mid-urethral stricture from his previous hypospadias repair, with a number of hairs present in the skin at the stricture site. The hairs were thought to be from graft skin used to repair his hypospadias, and this was felt to be the cause of his bleeding. He re-presented with intermittent urethral blood loss 2 years later, at 57 years of age, and repeat cystoscopy again showed a mid-urethral stricture that was treated by dilatation.
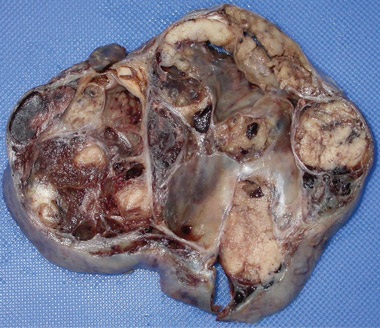
At 59 years of age, a laparotomy was performed and the patient was found to have a large left adnexal mass (Box 1, A) and a normally formed uterus. The mass was removed and a subtotal hysterectomy performed as there was no distinct lower margin of the cervix (Box 1, B). The patient was informed of the operative findings and the diagnosis of intersex. Peripheral blood karyotyping showed a 46,XY genotype. The histopathology of the gonadal tumour showed a sex cord-stromal tumour of indeterminate differentiation (Box 2). The histopathology of the uterus showed simple endometrial hyperplasia. The patient was treated with postoperative chemotherapy, but died 18 months later.
Discussion
Disorders of sex development (DSD) are rare in the general population, and comprise a wide variety of clinical, anatomical and genetic problems. Most patients with DSD are diagnosed at birth, but diagnosis may not be made until childhood, adolescence or, more rarely, in adulthood, as in our case. This case shows how incomplete initial assessment and treatment of a neonate with abnormal genital development, and failure to recognise this rare disorder in adulthood can have tragic, adverse health consequences.
An international consensus statement on intersex disorders and their management proposed the term “disorders of sex development” to describe congenital conditions with atypical development of chromosomal, gonadal and anatomical sex.1 It was considered that previous terminology, such as hermaphroditism, was controversial, pejorative to patients and confusing. DSD are a heterogeneous group of conditions, all of which interfere with normal sex differentiation in the embryo and fetus. The incidence of DSD in the population is estimated to be one in 5500.2
Patients with DSD can present at birth with ambiguous genitalia, apparent female or apparent male genitalia. Patients with apparent female genitalia can have an enlarged clitoris, posterior labial fusion or an inguinal / labial mass. Patients with apparent male genitalia can have non-palpable testes, micropenis, isolated perineal hypospadias or hypospadias with undescended testes. Congenital adrenal hyperplasia is the most common cause of ambiguous genitalia in the newborn period.2 The birth of an infant with ambiguous genitalia requires a strategy of clinical, hormonal, genetic, molecular and radiographic investigations to determine the aetiology and to plan a therapeutic approach.3 Clinical and diagnostic evaluation protocols are available.2
Although most DSD are diagnosed in the neonatal period, a wide variety of clinical presentations have been reported in childhood, adolescence and adulthood. Later presentations include previously unrecognised genital ambiguity, inguinal hernia in a girl, delayed or incomplete puberty, primary amenorrhoea, virilisation in a girl, breast development in a boy and gross haematuria in an adolescent male.1,2 A wide variety of clinical presentations have been reported in adults with undiagnosed DSD. These include psychiatric disturbances,4 short stature, infertility,4,5 lower abdominal pain6 or haematuria. Our patient presented with intermittent haematuria that was initially investigated with cystoscopy and an intravenous pyelogram. A computed tomography urogram with intravenous contrast has now replaced intravenous pyelography as the standard investigation of the urinary tract in patients with haematuria. If this investigation had been performed at the time of his earlier presentations, a pelvic mass may have been identified and further investigated.
Recent advances in molecular genetics have increased our understanding of both normal and abnormal sex development. The bipotential gonad develops at the urogenital ridge between 5 and 6 weeks’ gestation and a number of genes involved in testis determination are expressed at this stage.7 The first histological evidence of testis formation occurs at 7 weeks’ gestation. Leydig cell synthesis of testosterone mediates the development of the vas deferens, epididymis and seminal vesicles, and conversion of testosterone to its more potent form, dihydrotestosterone, masculinises the external genitalia to form a penis and scrotum. Sertoli cell synthesis of anti-Müllerian hormone prevents the Müllerian ducts from developing into a uterus and fallopian tubes.7,8 The development of internal reproductive organs is controlled by the ipsilateral gonad, and prenatal male sex development is finally completed at term by descent of the testes into the scrotum.7 Our patient appears to have produced enough testosterone from the small right testis to promote penile development, albeit with hypospadias. The left gonadal dysgenesis appears to have resulted in inadequate anti-Müllerian hormone production, allowing a uterus to develop.
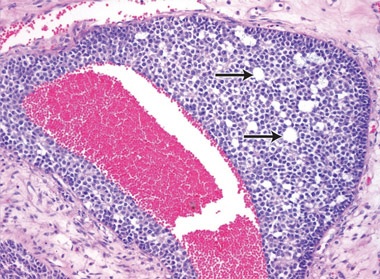
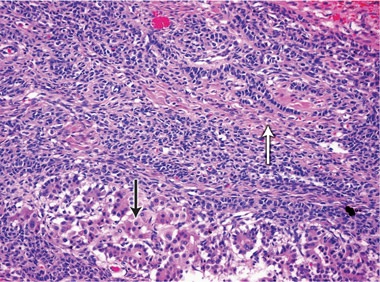
The risk of malignancy in dysgenetic gonads is significantly increased in some patients with DSD.8 The presence of the SPY gene on the GBY region of the Y chromosome is a prerequisite for malignant transformation.9 Tumours can arise in any of the gonadal cells or their precursors.8 Precursor lesions for the development of cancers occur as carcinoma-in-situ in testicular tissue and gonadoblastoma in undifferentiated gonadal tissue. A number of malignant tumour types may occur in dysgenetic gonads.8,10 These include germ cell tumours and sex cord-stromal tumours. Patients presenting with abdominal tumours in dysgenetic gonads in adulthood provide histopathologists with complex diagnostic dilemmas. Histopathological examination of the tumour in our case showed mixed elements, with cells resembling ovarian follicles, testicular tunica, granulosa cells, Sertoli cells and Leydig cells. No germ cell components were identified. The final consensus was a diagnosis of malignant sex cord-stromal tumour of indeterminate differentiation.
It is likely that the oestrogen-secreting gonadal tumour had been present for some years in our patient, resulting in endometrial hyperplasia. The uterus is likely to have been connected to the urethra, with endometrial bleeding causing the apparent haematuria. Persisting elevated levels of oestrogen would also have caused gynaecomastia, which resolved after surgical removal of the tumour. Serum follicle-stimulating hormone was likely to have been suppressed by the elevated level of oestradiol, and a normal level of serum luteinising hormone and low level of testosterone are consistent with the history of incomplete external genital development and infertility.
Clinicians managing neonates with cryptorchidism and proximal hypospadias should have a high index of suspicion of an underlying DSD.11 Once a DSD is identified, a series of investigations should be initiated with the aim of making a specific diagnosis.7,12 Physical examination and imaging studies should be performed to attempt to locate inguinal or abdominal gonads so that surgical removal can be considered in patients thought to be at high risk of subsequent malignancy.2,10 The sequence of events in our case highlights the diagnostic difficulties that can occur in patients who present in adulthood with unsuspected gonadal dysgenesis.
1 Macroscopic photographs of the left adnexal mass
 |
|
A: Macroscopic photograph of malignant sex cord-stromal tumourdisplaying widespread necrosis and haemorrhage. 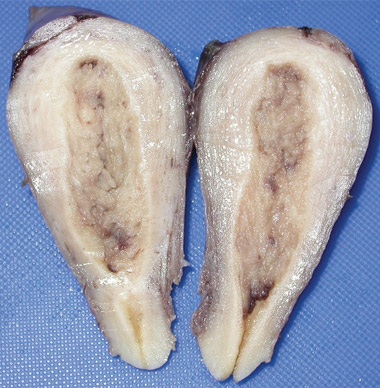 B: Macroscopic photograph of bisected uterus with a thickened endometrium. |
2 Histopathology of the gonadal tumour
Competing interests
None identified.
Acknowledgements
We thank Peter Russell of Douglass Hanly Moir Pathology for assisting with the interpretation of the histopathology results, and with the preparation of the microscopic illustrations.
References
- Houk CP, Hughes IA, Ahmed SF, Lee PA. Summary of consensus statement on intersex disorders and their management. Pediatrics 2006; 118: 753-757. 0_i1091956
- Nabhan ZM, Lee PA. Disorders of sex development. Curr Opin Obstet Gynecol 2007; 19: 440-445. 0_i1091958
- Sultan C, Paris F, Jeandel C, et al. Ambiguous genitalia in the newborn. Semin Reprod Med 2002; 20: 181-188. 0_i1091960
- Kurita M, Aiba E, Matsumoto D, et al. Feminizing genitoplasty for treatment of XX male with masculine genitalia. Plastic Reconstr Surg 2006; 117: 107-111. 0_i1091962
- Bostwick JM, Martin KA. A man’s brain in an ambiguous body: a case of mistaken gender identity. Am J Psychiatry 2007; 164: 1499-1505. 0_i1091964
- Lim AC, Stiel JN, Blome SA, Yip MY. A case of mistaken identity. A rare presentation of gonadal dysgenesis. Aust Fam Physician 2000; 29: 945-947. 0_i1091966
- Ahmed SF, Hughes IA. The genetics of male undermasculinization. Clin Endocrinol (Oxf) 2002; 56: 1-18. 0_i1091968
- Siowikowska-Hilczer J, Romer TE, Kula K. Neoplastic potential of germ cells in relation to disturbances of gonadal organogenesis and changes in karyotype. J Androl 2003; 24: 270-278. 0_i1091970
- Looijenga LH, Hersmus R, Oosterhuis JW, et al. Tumour risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 2007; 21: 480-495. 0_i1091972
- Stewart CJR, Baker E, Beaton C, et al. Detection of Y-chromosome in gonadal tumours using fluorescence in situ hybridization: diagnostic value in intersex conditions including older patients with clinically unsuspected androgen insensitivity syndrome. Histopathology 2008; 52: 175-182. 0_i1091974
- Marrocco G, Poscente M, Majore S, et al. Clinical management and molecular cytogenetic characterization in a 45,X/46,X,idic(Yp) patient with severe hypospadia. J Pediatr Surg 2003; 38: 1258-1262. 0_i1091976
- Robboy SJ, Jaubert F. Neoplasms and pathology of sexual developmental disorders (intersex). Pathology 2007; 39: 147-163. 0_i1091979