




Tumour necrosis factor inhibitors have revolutionised the management of Crohn’s disease, but reports of a possible association between concomitant infliximab and immunomodulator therapy and hepatosplenic T-cell lymphoma (a rare form of aggressive non-Hodgkin’s lymphoma) have emerged. We describe the first case in Australia of hepatosplenic T-cell lymphoma in a patient who had been treated with infliximab and immunomodulators for Crohn’s disease.
Abdominal computed tomography confirmed gross splenomegaly, with the spleen being 24 cm long on its major axis, but no lymphadenopathy. Microscopic examination of a liver core biopsy specimen revealed an atypical lymphoid infiltrate in the sinusoids, especially around central veins (Box), with immunophenotype bcl-2+, CD3+, CD43+, Ki67+, ALK1 −, bcl-6 − , CD5 −, CD10 −, CD20 −, CD30 −, CD79 − and cyclin D1 − . Subsequent genetic studies of a bone marrow biopsy specimen revealed rearrangement of the T-cell receptor γ-chain gene, consistent with hepatosplenic T-cell lymphoma (HSTCL).
The lymphoma was treated with one cycle of cyclophosphamide, mesna, dexamethasone, doxorubicin and vincristine, which achieved a partial response, but subsequent salvage chemotherapy with ifosfamide, carboplatin and etoposide did not arrest disease progression. In June 2006, the patient received a sibling allogeneic bone marrow transplant, after conditioning with etoposide and total body irradiation.1
We describe the first case, to our knowledge, in Australia of HSTCL in a patient with Crohn’s disease who had been treated with infliximab and immunomodulators. Inhibitors of tumour necrosis factor (TNF) such as infliximab have shown great efficacy in the treatment of luminal and fistulising Crohn’s disease, as well as ulcerative colitis.2 Infliximab is a chimeric (human/murine) monoclonal antibody that binds to human TNF-α. It was approved by the United States Food and Drug Administration (FDA) in 1998 for the treatment of moderate-to-severe Crohn’s disease when response to immunomodulator therapy is inadequate. More recently, the FDA has expanded the indication to include ulcerative colitis refractory to conventional treatment.
In Australia, the Pharmaceutical Benefits Advisory Committee recently approved TNF inhibitors for the treatment of Crohn’s disease. However, the safety of such biological therapy is a concern: a recent meta-analysis reported a threefold increase in the risk of malignancy (solid and haematological) with the use of anti-TNF therapy in rheumatoid arthritis patients.3 This increase may be partly due to severity of disease or to other aspects of disease management. Interestingly, to date there have been no reports of HSTCL in rheumatoid arthritis patients. The TREAT (Crohn’s Therapy Resource, Evaluation and Assessment Tool) registry, which monitors over 3000 patients with Crohn’s disease who have been treated with infliximab, has not reported an increased incidence of lymphoma.4 However, it is estimated that a substantially larger number of patients would need to be monitored to detect a significant increase in a rare adverse event, such as lymphoma.5 Between 2000 and 2006, the Adverse Drug Reactions Advisory Committee received 319 reports involving anti-TNF therapy, including five cases of lymphoma.6
HSTCL is a rare form of aggressive non-Hodgkin’s lymphoma that comprises 5% of peripheral T-cell lymphomas. Reports of approximately 120 cases have been published worldwide. Eight cases of HSTCL have been identified from the post-marketing infliximab safety database run by the FDA, which seems more than expected, all in young men with a history of Crohn’s disease and concomitant use of azathioprine or mercaptopurine.7 In addition, there have been 15 reports of ‘‘T-cell lymphoma’’ in patients treated with infliximab, but data from the Adverse Event Reporting System were limited.7 Furthermore, there have been no reports of HSTCL associated with other anti-TNF therapies (eg, etanercept and adalimumab) used for any indication.7 Of note, there have been four reports of HSTCL in patients who received azathioprine or mercaptopurine alone for 4–6 years.8
The case we describe differs substantially from previously reported cases of HSTCL associated with concomitant infliximab and immunomodulator treatment in Crohn’s disease. To our knowledge, it involves the oldest patient and longest lead time (72 months) reported to date. Patients in most other cases have been younger than 22 years, with a lead time of less than 58 months7 (unpublished data, Centocor, Horsham, Pa, USA).
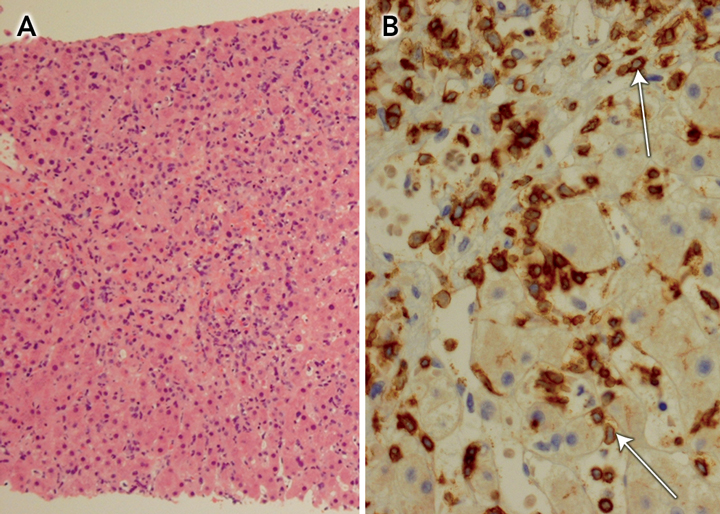
- 1. He S, Roberts A, Ritchie D, Grigg A. Graft-versus-lymphoma effect in progressive hepatosplenic gamma/delta T-cell lymphoma. Leuk Lymphoma 2007; 48: 1448-1450.
- 2. Osterman MT, Lichtenstein GR. Current and future anti-TNF therapy for inflammatory bowel disease. Curr Treat Options Gastroenterol 2007; 10: 195-207.
- 3. Bongartz T, Sutton AJ, Sweeting MJ, et al. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006; 295: 2275-2285.
- 4. Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol 2006; 4: 621-630.
- 5. Sackett DL. Bias in analytical research. J Chronic Dis 1979; 32: 51-63.
- 6. Tumor necrosis factor alpha inhibitors. Aust Adverse Drug React Bull 2006; 25 (6): 23.
- 7. Mackey AC, Green L, Liang LC, et al. Hepatosplenic T cell lymphoma associated with infliximab use in young patients treated for inflammatory bowel disease. J Pediatr Gastroenterol Nutr 2007; 44: 265-267.
- 8. Zeidan A, Sham R, Shapiro J, et al. Hepatosplenic T-cell lymphoma in a patient with Crohn's disease who received infliximab therapy. Leuk Lymphoma 2007; 48: 1410-1413.
None identified.