





Volume 189 - Issue 5
Chronic myeloid leukaemia: the evolution of gene-targeted therapy
Author: David J L Joske
Med J Aust 2008; 189 (5): 277-282. || doi: 10.5694/j.1326-5377.2008.tb02027.x
Published online: 1 September 2008
Published online: 1 September 2008
Abstract
Chronic myeloid leukaemia (CML) was the first human cancer linked to an acquired chromosomal abnormality, subsequently shown to be a reciprocal translocation between chromosomes 9 and 22. The resulting fusion gene product, BCR-ABL, was shown to be the causative agent of the disease.
CML has an incidence of around 1–2 cases per 100 000; in Australia, there are probably more than 200 new cases per year and more than 1300 prevalent cases.
Treatment of CML with imatinib has been a powerful vindication of the concept of rational, gene-targeted drug design.
Five-year published experience with imatinib at 400 mg orally daily demonstrates 89% overall survival and an estimated 93% freedom from disease progression. Adverse effects are mostly mild and transient.
Higher doses of imatinib may be more efficacious and will be studied in upcoming clinical trials in Australia; however, imatinib is almost certainly not curative.
Up to 28% of patients may have to stop imatinib because of intolerance or disease resistance, mostly due to point mutations of BCR-ABL. In this situation, many patients will respond to second- and third-generation tyrosine kinase inhibitors.
Management of CML patients should involve close monitoring, especially in the first 2 years, with regular cytogenetics and quantitative polymerase chain reaction to optimise response and identify suboptimal responders as early as possible.
Bone marrow transplantation remains the only known cure, but is reserved for patients whose kinase inhibitor therapy has failed, or who have advanced disease (accelerated phase or blastic transformation).
Chronic myeloid leukaemia (CML) has been the showcase disease for haematologists for nearly 50 years since the discovery of the Philadelphia chromosome (Ph) as the hallmark of the disease in 1960 by Nowell and Hungerford,1 — the first instance of an acquired chromosomal change linked with a human malignancy. In 1972, Rowley recognised this as a reciprocal translocation — the first such description — between chromosomes 9 and 22, juxtaposing the BCR and ABL genes onto the shortened derivative chromosome 22, producing a fusion protein with abnormal tyrosine kinase activity.2 BCR-ABL was identified in 19823 and, in 1990, cells transfected with BCR-ABL produced a myeloproliferative disorder typical of CML in a mouse model.4 Most recently, CML has been the first human malignancy treated with a gene-targeted therapy — imatinib — the principal subject of this review.
Incidence and natural history
CML has a similar incidence in all Western societies of around 1–2 cases per 100 000 population, with a peak incidence in the fifth decade. Excessive radiation exposure is the only recognised cause. In Australia, while the exact incidence is unknown, there are probably more than 200 new cases each year and over 1300 prevalent cases.
Before effective treatments became available, the disease pursued a unique course. The vast majority of patients are diagnosed in so-called chronic-phase CML, really a pre-leukaemia, characterised by fatigue, sometimes weight loss, splenomegaly, and a high white cell count consisting predominantly of mature neutrophils and promyelocytes. One-fifth of patients per year from diagnosis would enter the accelerated phase — a rise in white cell and platelet counts, basophilia, anaemia, and blast cells in the peripheral blood — and then transformation to blast crisis (ie, secondary acute leukaemia). Two-thirds of these are acute myeloid leukaemias, and one-third are acute lymphoblastic leukaemias, reflecting the stem-cell nature of the disease. These secondary acute leukaemias are notoriously refractory to treatment, with intensive chemotherapy producing transient or no remissions.
Diagnosis
The diagnosis of CML is best established with a bone marrow biopsy, which classically shows hypercellularity, normal morphology, and the presence of the Ph chromosome in most or all metaphases examined with cytogenetic techniques. Fluorescence in situ hybridisation (FISH) is a technique that can rapidly quantify the presence of the Ph chromosome in several hundred cells. Other chromosomal abnormalities, if found, mostly define cytogenetically evolved disease, a hallmark of transformation and, for this reason, traditional karyotype analysis should be done at diagnosis.
Evolution of therapies (Box 1)
Early therapies included busulfan, which was clumsy to titrate as well as leukaemogenic, and hydroxyurea, which is still frequently used initially to reduce a very high white cell count quickly (“cytoreduction”) to decrease the risk of organ damage from leukostasis. During the 1980s, interferon alfa was shown to reverse the proportion of leukaemic metaphases present in standard bone marrow cytogenetic analyses of patients with CML, producing two-thirds or complete clearance of Ph-positive metaphases (major cytogenetic response and complete cytogenetic remission [CCR], respectively) in 10%–38% of patients.5 However, interferon alfa was an expensive and unpleasant therapy, with almost universal lethargy and flu-like symptoms. More recently, advances in molecular biology have led to rational, targeted drug design and the development of imatinib, the first clinically successful tyrosine kinase inhibitor (TKI).
Monitoring disease response
During the 1990s, the great promises of the molecular biological revolution bore fruit, particularly in this disease. Rapid polymerase chain reaction (PCR) amplification of BCR-ABL RNA transcripts was developed,6 providing very sensitive quantitative assays (Q-PCR) of the amount of disease present.
Although assessment of the disease was traditionally performed with serial bone marrow cytogenetic studies, Q-PCR results are more informative, especially at low levels of disease (minimal residual disease). As with most cancers, clinical detection of CML usually occurs at a tumour burden of 1012 cancer cells; response can be defined as haematological (normalisation of the white cell count, which corresponds to about a 1-log reduction in the BCR-ABL Q-PCR), cytogenetic (reduction or disappearance of Ph-chromosome-positive marrow metaphases, or about 2-log reduction) or molecular (major molecular response equating to a 3-log reduction in the assay) (Box 2). Peripheral blood and bone marrow appear to give equivalent Q-PCR results.7
Introduction of imatinib
The late 1990s saw the introduction into clinical practice of imatinib mesylate (Glivec, Novartis; Gleevec in the United States), the first true “magic bullet” in cancer therapy — that is, a treatment that specifically interacts with abnormal cellular processes in cancer cells, largely sparing normal cellular processes. Imatinib is a synthetic TKI specifically designed to inhibit the BCR-ABL fusion protein, by competitive binding at the ATP-binding site.
The story of how Novartis (then Ciba-Geigy) was persuaded to embark on the very expensive process of bringing Glivec to the bedside has been told eloquently by the company’s chairman.8 The trade name Glivec was borrowed from an already registered name for another agent — a planned glioma vector (K Lynch, formerly Novartis Australia, personal communication).9 Initially, the compound was known as signal transduction inhibitor STI571 (although wags soon renamed it “stop taking interferon”).
Incredibly, early reports of the imatinib experience were collated by a CML patient who interviewed patients as they left the clinic and then presented this information online at a website.10 Rumours of the leukaemia melting away on four tablets a day rapidly escaped the trial centre in Oregon, USA. The pivotal IRIS (International Randomized Study of Interferon and STI571) trial between 2000 and 2001 randomly allocated 1106 patients to STI571 or standard care (then interferon alfa plus cytosine arabinoside, a purine antimetabolite). Crossover to STI571 was allowed for disease progression, intolerance of treatment, or failure to achieve a major cytogenetic response at 24 months. Progression was defined as death, accelerated phase, blast crisis, loss of major cytogenetic response (more than two-thirds of metaphases Ph-chromosome negative) and loss of complete haematological response. The landmark publication in 2003 of interim, very successful IRIS results heralded the era of gene-targeted cancer therapy11 and provided proof-of-concept.
Efficacy of imatinib
Five-year follow-up results from the IRIS trial allow a mature assessment of imatinib.12 A complete haematological response is seen in 97% of patients; and a complete cytogenetic response is seen in 82% of patients. The estimated 5-year progression-free survival is 84%, the estimated 5-year survival without progression to accelerated phase or blastic transformation is 93%, and the overall survival on first-line imatinib on an intention-to-treat analysis is 89.4%, rising to 95.4% if CML-unrelated deaths are excluded. Moreover, it is clear that patients who achieve major molecular response enjoy an extremely low risk of progression to accelerated or blastic phase CML, regardless of when the response is achieved. Another key finding is that this is maintained: there has been no increased incidence over time of progression to accelerated phase or blastic transformation, unlike with earlier therapies.
Safety of imatinib
Common toxicities include mild nausea, muscle cramps, and a distinctive peri-orbital oedema. Practical strategies to manage these are discussed in a recent review.13 Grade III or IV acute toxicities have been limited to myelosuppression, elevated liver enzyme levels and a handful of other drug-related adverse events. No unexpected long-term sequelae have emerged after 8 years. In a small subset of patients, imatinib may be associated with markers of increased and unbalanced bone remodelling.14
No increased cancer risk has yet been shown,15 but there are emerging reports of cytogenetic changes in the Ph-negative metaphases in up to 10% of Ph-negative cells, including trisomy 8, monosomy 7, and monosomy 5.16,17 Although frank myelodysplasia and even de novo acute myeloid leukaemia have been described,18 the prognosis is good if there are no obvious dysplastic morphological abnormalities, and therapy need not be altered.19
Ten cases of imatinib-associated cardiac failure were reported in 2006.20 This prompted reviews of the incidence in major CML centres. At the M D Anderson Cancer Center in Houston, Texas, 22 cases were identified among 1276 patients enrolled in imatinib clinical trials,21 a similar incidence to that found in the Framingham Heart Study. Of these, 18 had a recognised predisposition (eg, hypertension, diabetes) and 11 patients were able to continue on imatinib.
Imatinib is thought to have no effect upon developing sperm, but it is not recommended that men attempt to conceive while taking imatinib. Ninety-three such men have fathered children, with the outcome normal in all 41 known cases. In women, imatinib is teratogenic. There are reports of 180 pregnancies, including 71% with known first-trimester exposure. The outcomes are known for 125 cases: 63 had a normal infant, 35 women terminated, 18 (15%) had spontaneous abortion, and 12 (9.6%) had fetal abnormalities.22 Craniosynostosis, exomphalos, and hyperplastic kidneys were seen. The expected risk of abnormalities is about 10%.
Finally, there has been no evidence to date that imatinib has any deleterious effect on patients who proceed to allogeneic bone marrow transplantation (BMT), unlike interferon alfa. A recent Australian review discussed the role of allogeneic transplantation in the imatinib era.23 The authors superimposed survival curves from the IRIS trial results from the Center for International Blood and Bone Marrow Transplant Research for HLA-matched sibling allogeneic BMT in chronic phase. This showed superior results for imatinib at all time points up to 5 years from diagnosis. Nevertheless, allogeneic BMT remains the only curative approach and is still used in patients for whom TKIs fail or in advanced disease.
The success of imatinib is a powerful vindication of the concept of gene-targeted therapy. Imatinib has a dramatic effect on CML, with near-complete disease eradication in most patients, when assayed by the most sensitive techniques, and with a minimal side-effect profile.
That said, there is debate about the optimum dose of imatinib. A higher dose than the 400 mg used in the IRIS trial might differentiate between slow responders and those who ultimately prove to be non-responders. The Australian TIDEL I study demonstrated superior cytogenetic response rates using 600 mg/day.24 Box 3 shows a summary of clinical trials in CML. We await with interest the results of the TOPS trial in the US and Australia (comparing 400 mg/day with 800 mg/day) and the SPIRIT trial in Europe (comparing 400 mg/day with 600 mg/day). The proposed CML9 (TIDEL II) trial of the Australasian Leukaemia and Lymphoma Group uses escalating doses of imatinib if key milestones are not met at 3, 6 and 12 months; and prompts a change to a second-generation TKI (see below) at 18 months if required.
Can imatinib provide a cure?
Almost certainly not. BCR-ABL titres usually rise to baseline levels within months of stopping imatinib, even in patients with complete molecular remission.25 In one report of 12 patients in whom imatinib was discontinued after more than 2 years complete molecular remission, six patients remained Q-PCR negative after a median follow-up of 18 months.26 This contrasts with the experience from the M D Anderson Cancer Center. Among 10 women in whom imatinib was stopped early because of pregnancy, there were higher-than-expected rates of relapse and subsequent imatinib resistance.27 In these patients, prior imatinib exposure was relatively brief and full response had not been achieved. These observations fit with a model of biphasic effect of imatinib, with initial triggering of apoptosis in differentiated cells, followed by a slower decline associated with leukaemic stem-cell turnover (ie, longer remissions seen in those with depleted leukaemic stem cells).28 This latter effect is ultimately incomplete: leukaemic stem cells persist in low numbers, and thus single agent imatinib is unlikely to be curative. The TWISTER trial in Australia will study the effect of stopping imatinib in a small number of patients in long-term remission, with close molecular and clinical supervision (Box 3).
However, some patients either become intolerant of the drug, or their leukaemia develops resistance to it. The IRIS experience is that some 28% of patients will have to stop imatinib, principally for reasons of emerging disease resistance (about 20%) or of imatinib intolerance (8%). The peak incidence of imatinib failure is seen at about 2 years and seems to fall away after that time. The focus is now shifting to the management of these cases.
Mechanisms of imatinib resistance
Several mechanisms of resistance have been identified, but the commonest cause is mutation of the kinase domain, seen in 50%–75% of relapses. Since identification of the first mutation in 2001,29 at least 60 mutations have been described. A small number are seen frequently in more than 10% of patients; a lesser number are seen in 2%–10% of patients with mutations; and a number of others have been recognised in vitro or very rarely. These mutations directly affect tyrosine kinase binding to the molecule, but they may also alter substrate specificity and affect other signal transduction pathways. These mutations have been ranked in terms of their transforming potency upon cell lines, irrespective of their sensitivity to imatinib.30
For these reasons, mutation screening, detection and characterisation have become essential for optimal patient management. In Australia, mutation screening and detection is readily available because of the internationally recognised work of Hughes and Branford at the Institute of Medical and Veterinary Science and Royal Adelaide Hospital. Although mutation screening is not recommended at diagnosis (when the wild-type BCR-ABL clone invariably predominates), it should be done if there is emerging leukaemic resistance to imatinib (Box 4).
Second- and third-generation TKIs
The problem of resistance has led to the development of second- and now third-generation inhibitors. The two most studied presently are dasatinib (Sprycel, Bristol-Myers Squibb) and nilotinib (to be called Tasigna, Novartis; currently unregistered, but approved by the Australian Drug Evaluation Committee).
Dasatinib is 325 times more potent than imatinib at inhibiting BCR-ABL, at least in part because it binds to the active and inactive conformations of the molecule. It inhibits BCR-ABL and other tyrosine kinase processes, including C-Kit, PDGF-R, and SRC. Nilotinib is 20–50 times more potent than imatinib and inhibits BCR-ABL, C-Kit, and PDGF-R, but not SRC. Thus, only dasatinib inhibits SRC kinase, with the theoretical advantage that this pathway has been increasingly implicated in disease progression through mechanisms distinct from BCR-ABL.31
Phase 1 experience with both drugs has been presented.32,33 In chronic-phase CML, albeit with relatively early follow-up in both articles, complete cytogenetic responses were seen in around 35%–50% of imatinib-resistant patients with both agents; with updates on the initial reports, this figure will rise. Adverse events with nilotinib were mostly mild, and included raised bilirubin, itch, dry skin and rashes, but there are concerns over cardiac QT prolongation and three serious adverse cardiac events. Dasatinib was more myelosuppressive, and caused more diarrhoea, nausea and rashes. A unique side effect is pleural effusions in up to 17% of patients.
A Phase 2 randomised controlled trial of dasatinib has also been presented.34 Imatinib-resistant patients were randomly allocated to either 140 mg dasatinib (70 mg twice daily) or 800 mg imatinib (400 mg twice daily) in a 2 : 1 randomisation. At 15 months’ follow-up, superior results were seen in the dasatinib arm, with major cytogenetic responses in 52% versus 32% in the high-dose imatinib arm.
On present data, the efficacy of nilotinib and dasatinib seems broadly similar; in-vitro observations suggest that there are differences in the response of some of the known BCR-ABL point mutations to either drug. Mutation analysis may guide selection of a second-line agent. There remains at least one mutation — the so-called T315I — which confers resistance to imatinib, dasatinib and nilotinib, with no objective responses seen.
Fortunately, the third-generation TKIs are on the way. A handful of reports of patients treated with MK-0457 have been published.35 This is an aurora kinase inhibitor that affects spindle formation, inducing ineffective mitoses to undergo apoptosis. Of three patients with acute lymphoblastic leukaemia and the T315I mutation present, two achieved complete responses and there was one partial response. Other new kinase inhibitors in the pipeline include bosutinib (SKI-606), some 300 times more potent than imatinib, and experimental agents VX-680, BIRB-796, and ONO12380.
Treatment of advanced disease
Our understanding of what constitutes advanced disease is changing (eg, in patients who show acceleration defined by clonal evolution).36 But true accelerated phase and blast crisis patients remain a problematic patient group. Very briefly, imatinib (with chemotherapy mostly) initially produced encouraging responses, but they proved transient.37 In accelerated phase and blast crisis patients, 20%–40% are refractory to dasatinib and 30%–60% are refractory to nilotinib.32,33 Transplantation also gives unsatisfactory results, and new approaches are needed.
Optimal management of CML patients
Australasian guidelines have been formulated by the Australasian Leukaemia and Lymphoma Group (similar to recommendations proposed by Grigg and Hughes23) (Box 5); they are stricter than the European guidelines.39 Newly diagnosed CML patients in Australia and New Zealand should be assessed fully, including bone marrow biopsy at 0, 3, 6 and 12 months, looking for evidence of the Ph chromosome and other cytogenetic changes that might indicate accelerated or transforming disease. Imatinib should be commenced at 400 mg/day, or in higher doses as part of a clinical trial. All patients should be monitored by their molecular response to therapy, and in the CML9 (TIDEL II) proposed trial, patients who do not achieve satisfactory molecular responses at specified landmark times will be moved to escalated doses and even second-line therapy if necessary after inadequate response at 12 months. Management of advanced disease remains problematic.
Summary

Imatinib has proven to be nothing less than a stunningly effective and safe therapy, and it has not only heralded the era of gene-targeted therapy but also set the bar high. No serious long-term toxicities have emerged. Up to 20% of patients will become resistant with a peak incidence at 2 years, and around 10% will be intolerant. Strategies for these patients include increasing the dose of imatinib, or changing to second-generation TKIs. Mature trial results of higher-dose imatinib and further results from the newer TKIs are eagerly awaited. Nevertheless, most patients will achieve major molecular response when taking imatinib, and in these patients their average survival has been estimated recently at more than 20 years.40 With an eye to the future, an intriguing and very recent report canvasses the possibility that combination therapy with two or more TKIs, ultimately perhaps including an inhibitor of T315I mutation, might be the road to a cure.41
1 Currently available therapies for chronic myeloid leukaemia
2 Relationships between the putative number of leukaemic cells, response, and the level of BCR-ABL transcripts*
* This research was originally published in Blood: Baccarani et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2006; 108: 1809-1820. © American Society of Hematology.
3 Completed and current clinical trials in chronic myeloid leukaemia
Rapid escalation of imatinib dose; early change to nilotinib if molecular landmarks are not met |
Now accruing at many Australasian chronic myeloid leukaemia centres |
||||||||||||||
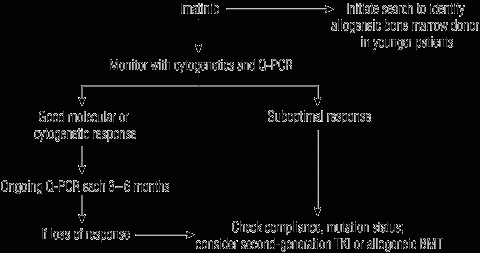
4 Suggested treatment algorithm for chronic myeloid leukaemia
 | |||||||||||||||
|
BMT = bone marrow transplantation. Q-PCR = quantitative polymerase chain reaction. TKI = tyrosine kinase inhibitor. Adapted from Grigg A, Hughes T,23 with permission of the authors and Biology of Blood and Marrow Transplantation. | |||||||||||||||
5 Australasian guidelines for management of chronic-phase chronic myeloid leukaemia
Initial therapy often includes hydroxyurea and allopurinol in patients with high tumour burden.
Imatinib 400 mg daily is the recommended starting dose unless patients are entering clinical trials of higher doses.
Initial monitoring should include weekly full blood count and attention to the risks of tumour lysis and myelosuppression. Adverse effects are usually mild, transient and manageable.13
Haematological toxicity should only rarely be a reason for dose interruption, even in the presence of severe thrombocytopenia (above 30 × 109/L is acceptable). Granulocyte colony-stimulating factor is usually effective in preventing neutropenia.
Close monitoring is important in the first 2 years, when most cases of resistant disease are detected. This should include cytogenetics at baseline, and 3, 6 and 12 months; and quarterly quantitative polymerase chain reaction (Q-PCR) at a laboratory that has validated its results using the standardised numerical international scale.38
Patients should achieve a 1-log reduction in BCR-ABL titre in each of Months 1–3, Months 4–6 and Months 7–12, or the cytogenetic equivalents (see Box 2).
If patients do not achieve these results (suboptimal responders), or if there is a subsequent loss of response with rising BCR-ABL titre, consideration should be given to assessing compliance, measuring imatinib serum levels, and performing mutation analysis.
For imatinib-resistant or intolerant patients, second-generation tyrosine kinase inhibitors or bone marrow transplantation should be considered. The choice of second-generation agent is complex and should be informed by awareness of mutation status and the likelihood of response to a particular drug.
The Australasian Leukaemia and Lymphoma Group CML9 (TIDEL II) trial is opening for accrual at an increasing number of Australasian centres, and clinicians are encouraged to participate. The trial dose starts at 600 mg and intensifies treatment if landmark responses are not met at 3, 6 and 12 months.
Competing interests
David Joske serves on the Novartis Australia Medical Advisory Board, and is a scientific advisor to the Novartis Global World CML Registry.
References
- Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science 1960; 132: 1497. 0_i1092132
- Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining [letter]. Nature 1973; 243: 290-293. 0_i1092134
- De Klein A, Geurts van Kessel A, Grosveld G, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myeloid leukemia. Nature 1982; 300: 290-293. 0_i1092136
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990; 247: 824-830. 0_i1092138
- The Italian Cooperative Study Group on Chronic Myeloid Leukemia. Interferon alfa-2a as compared with conventional chemotherapy for the treatment of chronic myeloid leukaemia. N Engl J Med 1994; 330: 820-825. 0_i1092140
- Cross NC, Feng L, Chase A, et al. Competitive polymerase chain reaction to estimate the number of BCR-ABL transcripts of chronic myeloid leukaemia patients after bone marrow transplantation. Blood 1993; 82: 1929-1936. 0_i1092142
- Ross DM, Branford S, Moore S, Hughes T. Limited clinical value of regular bone marrow cytogenetic analysis in imatinib-treated chronic phase CML patients monitored by RQ-PCR for BCR-ABL. Leukemia 2006; 20: 664-670. 0_i1092144
- Vasella D, Slater R. Magic cancer bullet: how a tiny orange pill is rewriting medical history. New York: Harper Business, 2003. 0_i1092146
- Gangloff JM. Novartis to lobby health plans to cover Glivec. CMLSupport.com, 2001. http://www.cmlsupport.com/cmlglivecreport0301.htm (accessed Jul 2008).
- Mayfield J. NEWCMLDRUG.COM [website]. http://www.newcml drug.com/ (accessed Jul 2008).
- O’Brien SG, Guilhot F, Larson RA, et al; IRIS Investigators. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348: 994-1004. 0_i1092154
- Druker BJ, Guilhot F, O’Brien SG, et al; IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408-2417. 0_i1092156
- Schiffer CA. BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. N Engl J Med 2007; 357: 258-265. 0_i1092158
- Berman E, Nicolaides M, Maki RG, et al. Altered bone and mineral metabolism in patients receiving imatinib mesylate. N Engl J Med 2006; 354: 2006-2013. 0_i1092160
- Pilot PR, Sablinska K, Owen S, Hatfield A. Epidemiological analysis of second primary malignancies in more than 9500 patients treated with imatinib. Leukemia 2006; 20: 148. 0_i1092162
- Medina J, Kantarjian H, Talpaz M, et al. Chromosomal abnormalities in Philadelphia chromosome-negative metaphases appearing during imatinib mesylate therapy in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase. Cancer 2003; 98: 1905-1911. 0_i1092164
- Terre C, Eclache V, Rousselot P, et al. Report of 34 patients with clonal chromosomal abnormalities in Philadelphia-negative cells during imatinib treatment of Philadelphia-positive chronic myeloid leukemia. Leukemia 2004; 18: 1340-1346. 0_i1092166
- Kovitz C, Kantargian H, Garcia-Manero G, et al. Myelodysplastic syndromes and acute leukaemia developing after imatinib mesylate therapy for chronic myeloid leukaemia. Blood 2006; 108: 2811-2813. 0_i1092168
- Deininger MW, Cortes J, Paquette R, et al. The prognosis for patients with chronic myeloid leukemia who have clonal cytogenetic abnormalities in Philadelphia-chromosome negative cells. Cancer 2007; 110: 1509-1519. 0_i1092170
- Kerkela R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 2006; 12: 908-916. 0_i1092172
- Atallah E, Durand JB, Kantarjian H, Cortes J. Congestive heart failure is a rare event in patients receiving imatinib therapy. Blood 2007; 110: 1233-1237. 0_i1092175
- Rosti G. Case studies: pregnancy. Reproductive health and imatinib therapy [oral presentation]. CML Global Opinion Leader Summit. Practical Management of CML 2007: new options, new hope; 2007 Mar 2–4; Athens, Greece. http://www.cmlgolscme.com (accessed Jun 2008).
- Grigg A, Hughes T. Role of allogeneic stem cell transplantation for adult chronic myeloid leukemia in the imatinib era. Biol Blood Marrow Transplant 2006; 12: 795-807. 0_i1092179
- Hughes T, Branford S, Reynolds J, et al. Higher dose imatinib (600 mg/day) with selective intensification in newly diagnosed CML patients in chronic phase; cytogenetic response rates at 12 months are superior to IRIS [abstract]. Blood 2002; 104: 1001. 0_i1092181
- Cortes J, O’Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood 2004; 104: 2204-2205. 0_i1092183
- Rousselot P, Huguet F, Rea D, et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood 2007; 109: 58-60. 0_i1092185
- Ault P, Kantarjian H, O’Brien S, et al. Pregnancy among patients with chronic myeloid leukemia treated with imatinib. J Clin Oncol 2006; 24: 1204-1208. 0_i1092187
- Sherbenou DW, Druker BJ. Applying the discovery of the Philadelphia chromosome. J Clin Invest 2007; 117: 2067-2074. 0_i1092189
- Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293: 876-880. 0_i1092191
- Griswold IJ, MacPartlin M, Bumm T, et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol 2006; 26: 6082-6093. 0_i1092193
- Warmuth M, Damoiseaux R, Liu Y, et al. SRC family kinases: potential targets for the treatment of human cancer and leukemia. Curr Pharm Des 2003; 9: 2043-2059. 0_i1092195
- Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354: 2531-2541. 0_i1092197
- Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 2006; 354: 2542-2551. 0_i1092199
- Kantarjian H, Pasquini R, Hamerschlak N, et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukaemia after failure of first-line imatinib: a randomized phase 2 trial. Blood 2007; 109: 5143-5150. 0_i1092201
- Giles FJ, Cortes J, Jones D, et al. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood 2007; 109: 500-502. 0_i1092203
- O’Dwyer ME, Mauro MJ, Kurilik G, et al. The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood 2002; 100: 1628-1633. 0_i1092205
- Sawyers CL, Hochhaus A, Feldman E, et al. Gleevec (imatinib mesylate) induces hematologic and cytogenetic responses in patients with chronic myeloid leukemia in myeloid blast crisis: results of a phase II study. Blood 2002; 99: 3530-3539. 0_i1092207
- Hughes T, Deininger M, Hochhaus A, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood 2006; 108: 28-37. 0_i1092212
- Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukaemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2006; 108: 1809-1820. 0_i1092213
- Anstrom KJ, Reed SD, Allen AS, et al. Long-term survival estimates for imatinib versus interferon-α plus low-dose cytarabine for patients with newly diagnosed chronic-phase chronic myeloid leukaemia. Cancer 2004; 101: 2584-2592. 0_i1092215
- O’Hare T, Eide CA, Deininger MW. BCR-ABL kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007; 110: 2242-2249. 0_i1092217