





Volume 188 - Issue 5
Prolonged absorption and delayed peak paracetamol concentration following poisoning with extended-release formulation
Authors: Darren M Roberts and Nicholas A Buckley
Med J Aust 2008; 188 (5): 310-311. || doi: 10.5694/j.1326-5377.2008.tb01629.x
Published online: 3 March 2008
Published online: 3 March 2008
A woman with acute poisoning from extended-release paracetamol presented at 14.5 hours post-ingestion. The paracetamol’s absorption phase and elimination half-life appeared prolonged, with peak blood concentration occurring at 20 hours post-ingestion, requiring an extended course of intravenous N-acetylcysteine. Current treatment recommendations, based on experience with a different formulation in the United States, may not be appropriate for the Australian formulation.
Clinical record
A 25-year-old woman, weighing 54 kg and with no other medical conditions, ingested 96 extended-release paracetamol tablets with suicidal intent (total dose, 64 g [1185 mg/kg]). Onset of nausea and intermittent vomiting occurred after a couple of hours, and about 9 hours later she reported the exposure to relatives. The woman was taken to a regional hospital, from where she was transferred to a tertiary hospital for management, arriving 14.5 hours post-ingestion.
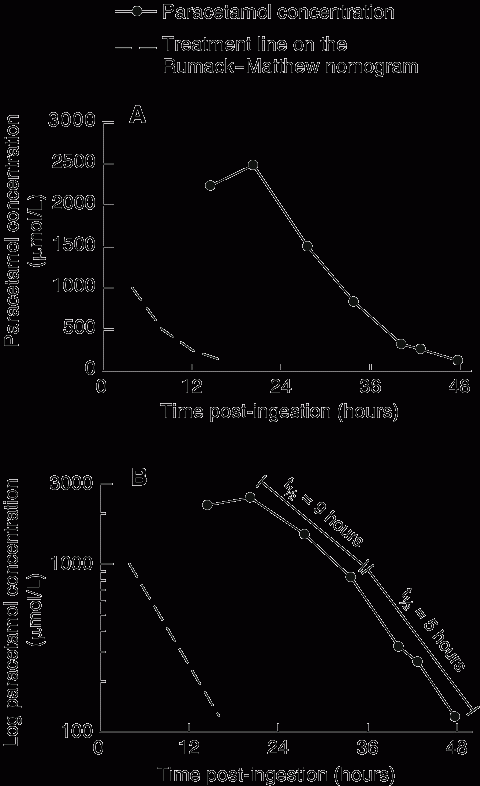
She was given antiemetics, and an intravenous N-acetylcysteine infusion was immediately commenced (standard regimen: 150 mg/kg over 60 min, then 50 mg/kg over 4 h, then 100 mg/kg over 16 h). A blood sample collected on admission for laboratory analyses showed a paracetamol concentration of 2235 μmol/L. As this concentration is above the treatment line on the Rumack–Matthew nomogram (Box, A), the N-acetylcysteine infusion was continued. Given the limited available data on the pharmacokinetics of extended-release preparations in overdose, blood samples were obtained about every 6 hours to guide treatment.
Changes in the blood paracetamol concentration during the patient’s stay are shown in the Box. The maximum concentration (2487 μmol/L) occurred at about 20 hours post-ingestion, and the apparent elimination phase appeared biphasic on visual inspection (Box, B). The apparent elimination half-life was 9 hours between 20 and 34 hours post-ingestion, decreasing to 5 hours between 34 and 48 hours post-ingestion, suggesting that absorption was ongoing during this time. With the exception of intermittent nausea and vomiting, the patient remained clinically well. After 48 hours, the paracetamol concentration had decreased to a non-toxic (therapeutic) concentration and results of liver function tests were normal. The N-acetylcysteine infusion was ceased, and the patient was transferred to a mental health unit.
Despite the large paracetamol ingestion and delayed initiation of N-acetylcysteine, no biochemical evidence of hepatotoxicity or other markers of significant toxicity were observed during admission. Mild coagulopathy was noted at 48 hours post-ingestion (prothrombin time, 19 s [reference range (RR), 9–15 s]; international normalised ratio, 1.7 [RR, 0.8–1.2]; activated partial thromboplastin time, 37 s [RR, 23–34 s]; fibrinogen, 1.3 g/L [RR, 1.5–4.0 g/L]), which is not expected to be significant in this clinical setting.1 A mild increase in total bilirubin level (peak, 28 μmol/L [RR, 2–20 μmol/L]) was also noted, but other liver function test results remained within reference ranges.
Discussion
Ingestion of paracetamol is one of the most common causes of acute intentional self-poisoning in Australia. Management is well described, and includes consideration of gastrointestinal decontamination and administration of intravenous N-acetylcysteine. The decision to administer N-acetylcysteine is based primarily on the plasma concentration of paracetamol and time of poisoning in relation to the treatment line on the Rumack–Matthew nomogram. Blood paracetamol concen-trations are obtained 4 or more hours after ingestion of immediate-release paracetamol preparations because absorption is largely complete by this time. Patients with a paracetamol concentration above the treatment line should be treated with this antidote, while those with a level below the line do not require treatment.2 N-acetylcysteine is administered as a 20-hour intravenous infusion in the majority of patients. These treatment guidelines reflect clinical experience with the standard immediate-release formulation of paracetamol, with or without coformulants such as codeine or dextropropoxyphene.
An extended-release formulation of paracetamol was recently marketed in Australia under three proprietary labels. Each tablet contains 665 mg paracetamol; 31% of the dose is released immediately, while the remaining 69% is released over 6–8 hours at therapeutic doses.3
To determine whether N-acetylcysteine is required in an acute overdose of an extended-release formulation, the manufacturer recommends obtaining an initial paracetamol concentration either on admission or at 4 hours post-ingestion. If this concentration is below the treatment line on the nomogram, a repeat concentration should be determined 4–6 hours later. A 20-hour infusion of N-acetylcysteine is recommended if either concentration is above the treatment line.3
This approach to risk assessment is similar to that recommended for a different extended-release preparation marketed in the United States (650 mg paracetamol; 50% released immediately).4 In volunteer studies of simulated overdose with this US formulation (75 mg/kg orally), the time of peak concentration was similar to the immediate-release formulation, occurring within 4 hours.5,6 However, a study of simulated acute overdose in volunteers using the Australian product (mean dose, 73 mg/kg) found that the absorption phase is prolonged — compared with the standard immediate-release formulation, the time until the maximum concentration occurred was delayed for the extended-release formulation (0.94 v 2.83 hours).7 These differences in absorption kinetics between the Australian and US formulations might influence treatment guidelines.
Reported cases of acute overdose with the US formulation have noted that the peak concentration may actually occur up to 12 hours post-ingestion, suggesting that the pharmacokinetics of extended-release paracetamol change substantially in acute overdose.8-12 To our knowledge, no cases of acute poisoning with the Australian formulation of extended-release paracetamol have previously been reported. It is therefore not known whether the current recommendations for treatment are appropriate.
Absorption may be sufficiently prolonged in acute overdose of extended-release paracetamol that the peak plasma concentration occurs around 20 hours post-ingestion. In this patient’s case, the only data point earlier than this was at 14.5 hours, when the concentration was already extremely high (Box). Given the amount ingested, it seems likely that at least one of the blood samples that the manufacturer recommends obtaining within 8–10 hours of ingestion would have prompted treatment with N-acetylcysteine. However, due to the lack of data during this period for our patient, this cannot be confirmed.
Of importance in this case is the prolonged apparent elimination half-life of paracetamol, such that the concentration remained elevated for 48 hours post-ingestion. This required the N-acetylcysteine infusion to be administered beyond the usual 20-hour protocol that is recommended by the manufacturer and used for immediate-release preparations. The duration of N-acetylcysteine infusion in this patient was guided by the serial blood samples taken about every 6 hours. In the absence of more informative pharmacokinetic data, it seems reasonable that serial blood samples (every 6–12 hours) should be obtained from patients presenting with ingestion of the extended-release formulation, to guide duration of treatment.
The absorption kinetics observed in this patient suggest that the ingested tablets formed an aggregate (a pharmacobezoar) in the gut from which absorption is very slow. Therefore, administration of activated charcoal and, potentially, whole bowel irrigation might be considered, even in cases of delayed presentation.13
On the basis of this case, the treatment of acute poisoning with the extended-release formulation of paracetamol differs from recommendations developed for the immediate-release formulation. While it appears reasonable to continue to use the Rumack–Matthew nomogram for determining which patients require N-acetylcysteine, more than one blood sample may be required to confirm that an exposure is non-toxic. Intravenous N-acetylcysteine infusion can be initiated according to the standard regimen, and should be continued until the paracetamol concentration has decreased to a therapeutic level (less than 120 μmol/L or 20 mg/L) if the transaminase concentrations remain normal. If the transaminase levels are rising, N-acetylcysteine should be continued. This can be determined by blood samples taken every 6–12 hours, depending on the time of day, location of the patient, laboratory services available, and compliance of the patient. If the infusion is required for longer than 20 hours, this should be done by continuing the final phase of the regimen (ie, N-acetylcysteine 100 mg/kg bodyweight in 1000 mL of 5% dextrose over 16 hours) until a therapeutic paracetamol concentration is achieved.
More experience in the management of acute poisoning with the extended-release formulation of paracetamol is required to better determine the optimal treatment.
Linear (A) and logarithmic (B) paracetamol concentration–time profiles after ingestion of extended-release paracetamol, with reference to the treatment line on the Rumack–Matthew nomogram
 | |||||||||||||||
|
t1/2 = apparent elimination half-life (determined using non-linear regression with a monophasic decay, with GraphPad Prism, version 4.03 for Windows [GraphPad Software, San Diego, Calif, USA]). | |||||||||||||||
Competing interests
None identified.
References
- Whyte IM, Buckley NA, Reith DM, et al. Acetaminophen causes an increased international normalized ratio by reducing functional factor VII. Ther Drug Monit 2000; 22: 742-748. 0_i1091868
- Rumack BH. Acetaminophen hepatotoxicity: the first 35 years. J Toxicol Clin Toxicol 2002; 40: 3-20. 0_i1091870
- eMIMS. Sydney: CMPMedica Australia, 2007. http://www.mims.com.au (accessed Feb 2008).
- Temple AR, Mrazik TJ. More on extended-release acetaminophen. N Engl J Med 1995; 333: 1508-1509. 0_i1091874
- Stork CM, Rees S, Howland MA, et al. Pharmacokinetics of extended relief vs regular release Tylenol in simulated human overdose. J Toxicol Clin Toxicol 1996; 34: 157-162. 0_i1091876
- Douglas DR, Sholar JB, Smilkstein MJ. A pharmacokinetic comparison of acetaminophen products (Tylenol Extended Relief vs regular Tylenol). Acad Emerg Med 1996; 3: 740-744. 0_i1091878
- Tan C, Graudins A. Comparative pharmacokinetics of Panadol Extend and immediate-release paracetamol in a simulated overdose model. Emerg Med Australas 2006; 18: 398-403. 0_i1091880
- Cetaruk EW, Dart RC, Hurlbut KM, et al. Tylenol Extended Relief overdose. Ann Emerg Med 1997; 30: 104-108. 0_i1091882
- Cetaruk EW, Dart RC, Horowitz RS, Hurlbut KM. Extended-release acetaminophen overdose. JAMA 1996; 275: 686. 0_pgfId-1091884
- Ho SY, Arellano M, Zolkowski-Wynne J. Delayed increase in acetaminophen concentration after Tylenol PM overdose. Am J Emerg Med 1999; 17: 315-317. 0_pgfId-1091885
- Vassallo S, Khan AN, Howland MA. Use of the Rumack–Matthew nomogram in cases of extended-release acetaminophen toxicity. Ann Intern Med 1996; 125: 940. 0_pgfId-1091886
- Bizovi KE, Aks SE, Paloucek F, et al. Late increase in acetaminophen concentration after overdose of Tylenol Extended Relief. Ann Emerg Med 1996; 28: 549-551. 0_i1091887
- Buckley NA, Dawson AH, Reith DA. Controlled release drugs in overdose. Clinical considerations. Drug Saf 1995; 12: 73-84. 0_i1091890