





Volume 186 - Issue 8
Fatal late-onset ornithine transcarbamylase deficiency after coronary artery bypass surgery
Authors: Mary Anne Chiong, Bruce H Bennetts, Simone I Strasser and Bridget Wilcken
Med J Aust 2007; 186 (8): 418-419. || doi: 10.5694/j.1326-5377.2007.tb00976.x
Published online: 16 April 2007
Published online: 16 April 2007
Clinical record
A 44-year-old man underwent coronary artery bypass surgery in 2004. He had been in satisfactory health as an adult apart from hypertension, for which he was receiving treatment. He ate a normal diet, including dairy products, meat and other high protein food. At age 44 years, he developed acute central chest pain while exercising at a gymnasium. An angiogram showed coronary artery occlusions.
Forty-eight hours after successful coronary artery bypass surgery, during which time he received intravenous sodium but not glucose, he felt unwell, and the following day he became delirious. Computed tomography of the brain, initially reported as appearing normal, was later thought to show cerebral oedema. Plasma ammonium level was 110 μmol/L (reference range [RR], 10–50 μmol/L), but the timing of this sample was unclear. Wilson’s disease was initially considered because of elevated liver enzyme levels, but serum copper and ceruloplasmin levels were not measured.
Eight days after the bypass surgery, a urine sample was sent to the NSW Biochemical Genetics Service, for metabolic screening. This showed gross elevations in glutamine and orotic acid (orotic acid, 14.8 μmol/mmol creatinine; RR, 1.23 μmol/mmol creatinine), indicating a likely diagnosis of ornithine transcarbamylase (OTC) deficiency. Plasma glutamine level was 3527 μmol/L (RR, 385–862 μmol/L). Plasma tyrosine and methionine levels were also moderately elevated, which was consistent with liver dysfunction. Plasma citrulline level was mildly elevated at 57 μmol/L (RR, 10–45 μmol/L), but this finding was difficult to interpret in the light of the other elevated amino acid levels. (Citrulline level is very low in neonatal OTC deficiency, but not necessarily low in late-onset phenotypes.)
The patient was by then gravely ill, requiring maximum life support, and under consideration for liver transplantation. His condition progressively deteriorated despite introduction of intravenous sodium benzoate and l-arginine, and life support was withdrawn. He died 8 days after surgery.
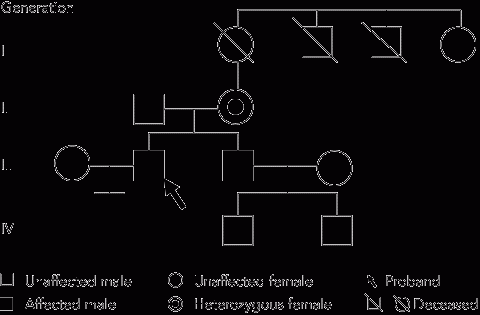
The diagnosis of OTC deficiency was subsequently confirmed by mutation analysis which showed hemizygosity for the c.622G>A (p.A208T) mutation in exon 6 of the OTC gene (as males have only one X chromosome, they are said to be hemizygous with respect to X-linked genes). Cascade testing of family members (Figure) showed that the patient’s mother (person II-2) was a carrier, heterozygous for the c.622G>A (p.A208T) mutation, while his asymptomatic brother (III-2) was hemizygous for the same mutation.
Random biochemical testing of the brother showed normal levels of plasma glutamine, citrulline, arginine and urinary orotic acid. No information was available on causes of death of first-generation relatives.
Past history revealed that, at age 7 years, the patient had an episode of acute encephalopathy following a 2-month history of intermittent nausea, vomiting and frontal headache. There was no preceding febrile illness, intercurrent infection or history of trauma. The symptoms reappeared after a 2-week period of apparent recovery. On admission to hospital, he was drowsy, disorientated and restless, but quickly settled and made a satisfactory recovery. No haematological or biochemical results were evident in his medical records. He was discharged with a diagnosis of viral meningitis/encephalitis.
We report a 44-year-old man who presented with fatal hyperammonaemia after coronary artery bypass surgery. He had previously been asymptomatic, apart from a possible episode of unrecognised hyperammonaemia in childhood. The diagnosis of ornithine transcarbamylase (OTC) deficiency was made too late for successful intervention. Inborn errors of metabolism are frequently unrecognised or diagnosed late in adults.
OTC deficiency is the most common disorder affecting the urea cycle. In New South Wales, the incidence is of the order of one in 70 000 births.1 It is an X-linked disorder leading to potentially lethal hyperammonaemia. The clinical severity ranges from acute neonatal hyperammonaemic coma to symptom onset at any time from infancy to adulthood, depending on environmental triggers and residual enzyme activity.2 The timing of episodes in late-onset OTC is dictated by environmental factors that increase nitrogen turnover, including dramatic increase in protein intake, medications affecting protein catabolism, viral illness or other generalised stress, rapid weight loss, and poor nutritional intake.3
Lessons from practice
Inborn errors of metabolism are frequently unrecognised or diagnosed late in adults.
Postoperative catabolism with insufficient calorie intake may unmask previously asymptomatic, but potentially lethal, inborn metabolic errors.
Patients with acute onset of unexplained neurological or psychiatric symptoms need urgent metabolic investigation, including measurement of plasma ammonia level, to exclude metabolic causes.
The first step in ureagenesis is the production of carbamyl phosphate from ammonium and bicarbonate. OTC then catalyses the biosynthesis of citrulline from ornithine and carbamyl phosphate. Thus, a deficiency of OTC leads to accumulation of ammonia and glutamine (the major extrahepatic source of ammonia for ureagenesis), and a reduction in citrulline. The accumulating carbamyl phosphate enters the pyrimidine synthetic pathway, resulting in increased excretion of orotic acid.
The missense mutation p.A208T, replacing alanine with threonine at codon 208 of exon 6 in the OTC gene, has been previously reported in a late-onset OTC patient we investigated,4 and in others.5-7 This group showed an extremely wide phenotype, ranging from encephalopathy at age 4 months7 to no symptoms at age 97 years.5
Our patient developed hyperammonaemia following postoperative catabolism, caused by surgical stress and fasting with inadequate calorie supply from intravenous fluids pre- and postoperatively. In addition, a high nitrogen load from bleeding sites, tissue trauma or tissue protein breakdown could have overwhelmed urea synthesis and promoted the excessive ammonia production. OTC deficiency is not the only inborn error of metabolism that can result in fatal postoperative decompensation. Another example is medium-chain acyl-CoA dehydrogenase deficiency, which appears more prevalent, with several recorded cases (eg, Raymond et al8). Other mild fatty-acid oxidation defects and, perhaps, intermittent maple syrup urine disease could behave similarly.
Death during an initial episode seems frequent in patients with late-onset OTC deficiency, as lack of familiarity with the disorder in the adult setting delays diagnosis and appropriate treatment.9-11 Treatment of hyperammonaemia is well established, and includes aggressive calorie support to counteract catabolism, early use of intravenous sodium benzoate as an ammonia sink, and intravenous arginine.2 It was unfortunate that no objective evidence was gathered on the cause of the patient’s episode of encephalopathy during childhood. While the cause could have been viral encephalitis, the clinical course, and appearance of the cerebrospinal fluid on microscopy and the brain on computed tomography did not strongly support this diagnosis. Assessment of plasma ammonia level would most likely have led to the diagnosis of OTC deficiency.
Establishment of the correct diagnosis in the patient led to the finding that his mother and brother were also affected, enabling them to be advised about precautions. The genetic implications for the family’s younger generation were not critical, as the proband had no children, and his brother’s children were both male and could not have inherited their father’s X chromosome (Figure).
This case illustrates the difficulty of diagnosing late-onset OTC. The X-linked inheritance may be obscured, even when more than one family member is affected, as some patients remain asymptomatic. Patients with acute onset of unexplained neurological or psychiatric symptoms need urgent metabolic investigation, including measurement of plasma ammonia level, to exclude metabolic causes.
Cascade testing of family members
Competing interests
None identified.
Acknowledgements
We thank Dr Kevin Carpenter (NSW Biochemical Genetics Service, Sydney, NSW) for the biochemical analysis and for editorial comments on the manuscript.
References
- Wilcken B. Problems in the management of urea cycle disorders. Mol Genet Metabol 2004; 81 Suppl 1: S86-S91. 0_i1091843
- Brusilow SW, Horwich AL. Urea cycle enzymes. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill, 2001: 1909-1964. 0_i1091845
- Summar ML, Barr F, Dawling S, et al. Unmasked adult-onset urea cycle disorders in the critical care setting. Crit Care Clin 2005; 21 (4 Suppl): S1-S8. 0_i1091847
- Ellaway C, Bennetts B, Tuck R, Wilcken B. Clumsiness, confusion, coma and valproate. Lancet 1999; 353: 1408. 0_i1091849
- Ausems M, Bakker E, Berger R, et al. Asymptomatic and late-onset ornithine transcarbamylase deficiency caused by a A208T mutation: clinical, biochemical and DNA analyses in a four-generation family. Am J Med Genet 1997; 68: 236-239. 0_CHDDEIEF
- Tuchman M, Morizono H, Rajagopal BS, et al. Identification of “private” mutations in patients with ornithine transcarbamylase deficiency. J Inher Metab Dis 1997; 20: 525-527. 0_pgfId-1094359
- Bakker E, Ausems M, Zaremba J, et al. Late onset and asymptomatic ornithine transcarbamylase deficiency (OTCD) caused by an A208T mutation [abstract]. Am J Hum Genet 1995; 57: A235. 0_i1091853
- Raymond K, Bale AE, Barnes CA, Rinaldo P. Medium-chain acyl-CoA dehydrogenase deficiency: sudden and unexpected death of a 45 year old woman. Genet Med 1999; 1: 293-294. 0_i1091855
- Finkestein J, Hauser E, Leonard C, Brusilow S. Late onset ornithine transcarbamylase deficiency in male patients. J Pediatr 1990; 117: 897-902. 0_i1091857
- Thakur V, Rupar CA, Ramsay DA, et al. Fatal cerebral edema from late-onset ornithine transcarbamylase deficiency in a juvenile male patient receiving valproic acid. Pediatr Crit Care Med 2006; 7: 273-276. 0_pgfId-1094390
- Rohininath T, Costello DJ, Lynch T, et al. Fatal presentation of ornithine transcarbamylase deficiency in a 62-year-old man and family studies. J Inherit Metab Dis 2004; 27: 285-288. 0_i1091862