




In Western countries, we are seeing the waning of a prolonged epidemic of peptic ulcer (PU) that began in the 19th century. In the 1960s, Susser and Stein first drew attention to the strange temporal behaviour of PU mortality.1 In the early 19th century, there was a high prevalence, in young women of the servant class, of acute gastric ulcer (GU) that tended to bleed or perforate at the gastric cardia.2 Remarkably, at the turn of the 19th century, this phenomenon disappeared, but was followed by an increase in the incidence of perforated juxtapyloric ulcers, initially in young and middle-aged men and later in older men. Extensive official data show that in the United Kingdom there was a plateau and then a decline in GU mortality and, after a lag of about 5 years, in duodenal ulcer (DU) mortality. In England and Wales, the risk of ulcer appeared in people born in the middle of the 19th century, with GU and DU reaching their peak in those born around 1885 and 1890, respectively.3 The risk of death from ulcer rose with age for both sexes, peaking in those born in the late 19th century and then falling steadily to current vanishingly small rates in young people in contemporary Western society (Box).
The geography of these phenomena is intriguing, with a similar cohort effect demonstrated in Japan,4 Switzerland,5 New York,6 Australia7 and several European countries.8 There are also data on social class differences. For England and Wales, data available from 1921 show that mortality rates for both GU and DU fell (in younger age groups) with increase in socioeconomic status, but that the trend reversed in old age. Susser and Stein speculated on whether this was related to the early phases of urbanisation or nutritional factors in the UK at the turn of the century,1 but similar data from Switzerland and Japan, despite their different social conditions, make these explanations unlikely. The possibility that the fall in prevalence of PU was the result of better treatment has been discounted by the long history of use of bismuth preparations for treatment of dyspepsia and their known inhibitory effect on Helicobacter pylori.9
In seeking a unifying explanation, the major factors to be examined are H. pylori, non-steroidal anti-inflammatory drugs (NSAIDs), smoking, stress, alcohol, dietary factors, physical activity and heredity.
Since the Nobel Prize-winning demonstration of the role of H. pylori in PU, there has been a widespread assumption that H. pylori is the cause of PU and so implicitly of the epidemic.10 Baron and Sonnenberg11 have suggested that an epidemic of a potent strain of H. pylori sweeping the world was the cause of the ulcer epidemic, and there are temporal, geographic and theoretical arguments to support this. While there may be reluctance to accept the potent H. pylori strain explanation, a birth cohort phenomenon in tuberculosis12 shows fascinating parallels to the time course of PU: “A plethora of studies . . . implicates some sort of generation effect in earlier life experience.”13 This fits neatly into the concept of the childhood acquisition of H. pylori leading to an epidemic of PU.14 Other examples of epidemics of infective disease sweeping the planet include diphtheria (from 1858) and scarlet fever a few decades later.15
The data show that people infected with H. pylori have a relative risk (RR) of 1.6–5.7 (mean, 3.3) of developing PU compared with non-infected people.16-18 Calculation of the population attributable risk (PAR) (ie, the proportion of disease cases that can be attributed to a particular factor) shows that, in recent series, H. pylori accounts for only about 50% of PU.19 This is consistent with the evidence reviewed below for other contributing factors. Even if the original pandemic strain were more pathogenic, with, for example, 90% of the population infected and an odds ratio (OR) of 5.0 of developing PU, the PAR (0.78) would still fall short of 1.00, leaving a role for factors such as smoking, which was very prevalent during the same time period.
There is evidence that smoking increases the risk of PU. An OR of 2.2 is typical, and a 40-year study of male doctors in the UK has shown a threefold increase in the risk of death from PU in current smokers.20
Until the mid 19th century, smoking of coarse pipe tobacco was usual, but about that time, a massive increase in cigarette smoking began.21 British soldiers returning from the Crimea had seen their Turkish allies using cigarettes; tobacco became more affordable; newer curing methods produced milder tobacco; and, in the United States, Buck Duke used advertising, developed packaging and improved cigarette-making machinery so that cigarettes could be produced cheaply. All wars have led to an increase in cigarette consumption — clearly evident by World War I. In the US, UK, Germany and France, per capita consumption rose between 1920 and 1965, then declined.22 While these facts support a role for smoking in ulcer epidemics, they ignore one important finding. The rise in tobacco use was largely confined to men. Not until their partial liberation after World War I could women smoke and retain their societal reputation. Yet the PU epidemic affected both men and women. Moreover, in the late 20th century, smoking in women increased while ulcer prevalence fell.
There is little evidence of an association between alcohol use and PU. A study of UK doctors by Doll et al found no association in 13 years of follow-up.23
Given the data and the time course of the PU epidemic, there is little in favour of caffeine as a causative factor in PU.25
The sparse evidence for a link between stress and PU is beset with methodological problems. A follow-up of 5388 people in the first US National Health and Nutrition Examination Survey (1986–1992) found a clear positive dose–response curve. Those with the highest level of stress had an adjusted RR of 2.9 (95% CI, 1.2–7.5) of developing PU.28
On balance, we can accept a link between stress and PU, but it does not really explain the epidemic.
There is a link between physical activity and PU, and the coincident fall in PU prevalence and in heavy physical work in Western countries typifies this. However, the long-term time trends of occupational workload and its underlying cohort pattern are different from those of PU.29
A role for hereditary factors in PU is clearly delineated by several Scandinavian studies of large groups of monozygotic and dizygotic twins, reared together and separately. The evidence indicates that 39%–62% of the susceptibility to PU is explained by hereditary factors, the rest by environmental ones.30,31 There is also a major role for heredity in H. pylori acquisition (63% attributable to genetic factors), but genetic influences for developing PU are independent of those for acquiring H. pylori, and the transition of H. pylori positivity to PU is due to environmental factors.32 There are two significant implications of these data. Firstly, the sum of the PARs for PU (the joint effects of heredity on PU and H. pylori colonisation) exceeds 1.00. Secondly, it is only through these factors that heredity can be considered as having a role in the PU epidemic.
We are left with very inadequate explanations for the waning epidemic of PU in recent years. Why young women of the servant class in the first half of the 19th century should have developed gastric ulcers is a total mystery. The next epidemic, beginning in cohorts born before 1885 and with a 5–10-year gap between GU and DU, is nearly as mysterious. Both H. pylori and cigarette smoking can claim a major responsibility in terms of timing. Neither, however, explains the rise in ulcer prevalence followed by a steady decline in prevalence and mortality rates while H. pylori colonisation rates remained high in the elderly.
A change in the H. pylori–human relationship along the lines of the Mycobacterium tuberculosis–human relationship may play a role. Certainly, while we know that M. tuberculosis is essential to development of tuberculosis, other factors are involved. There is overwhelming evidence that its major decline in Western society pre-dates and has been little influenced by medical intervention.33
The use of aspirin or other NSAIDs can not account for the PU epidemic, as they were not available in the 19th century, when the epidemic started.
While the beginning of the epidemic of PU in Western societies could have been caused by the coincidence of an epidemic of a highly pathogenic strain of H. pylori and the rapid growth of tobacco smoking, the subsequent course of the epidemic does not favour this simple hypothesis. An increase in tobacco smoking in the US between 1920 and 1960 occurred at the same time as a fall in incidence of PU. An alternative explanation is a gradual fall in the pathogenicity of H. pylori during the 20th century, similar to the apparent fall in the pathogenicity of organisms such as those causing scarlet fever and tuberculosis, both quite unrelated to the development of antibiotics.
In summary, the best hypothesis at present is that there has been an epidemic of PU at least partly due to the combination of H. pylori and cigarette smoking and that it will disappear from the list of important gastrointestinal disorders before we fully determine its cause.
Standardised cohort mortality ratio of gastric ulcer (GU) and duodenal ulcer (DU) analysed separately for men and women from England and Wales*
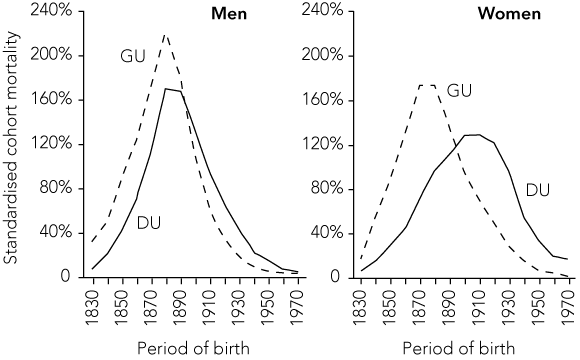
* From Sonnenberg A. A personal history of giving birth to the cohort phenomenon of peptic ulcer disease. In: Marshall B, editor. Helicobacter pioneers. Singapore: Blackwell Science Asia, 2002: 56. Reproduced with permission from Blackwell Publishing.
- John M Duggan1
- Anne E Duggan2
- 1 Princeton Medical Centre, Newcastle, NSW.
- 2 Gastroenterology Department, John Hunter Hospital, Newcastle, NSW.
None identified.
- 1. Susser M, Stein Z. Civilization and peptic ulcer. Lancet 1962; 1: 115-119.
- 2. Ellis H. A history of surgery. London: Greenwich Medical Media Limited, 2001: 114.
- 3. Susser M. Period effects, generation effects and age effects in peptic ulcer mortality. J Chronic Dis 1982; 35: 29-40.
- 4. Sonnenberg A, Muller A. Cohort and period effects in peptic ulcer mortality from Japan. J Chronic Dis 1984; 37: 699-704.
- 5. Sonnenberg A. Occurrence of a cohort phenomenon in peptic ulcer mortality from Switzerland. Gastroenterology 1984; 86: 398-401.
- 6. Baron JH, Sonnenberg A. Period – and cohort – age contours of deaths from gastric and duodenal ulcer in New York 1804–1998. Am J Gastroenterol 2001; 96: 2887-2891.
- 7. Westbrook JI, Rushworth RL. The epidemiology of peptic ulcer mortality 1953–1989: a birth cohort analysis. Int J Epidemiol 1993; 22: 1085-1092.
- 8. Sonnenberg A, Muller H, Pace F. Birth-cohort analysis of peptic ulcer mortality in Europe. J Chronic Dis 1985; 38: 309-317.
- 9. Marshall B. Commentary: Helicobacter as the “environmental factor” in Susser and Stein’s cohort study of peptic ulcer disease. Int J Epidemiol 2002; 31: 21-22.
- 10. Marshall BJ, Warren R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulcer. Lancet 1984; 1: 1311-1315.
- 11. Baron JH, Sonnenberg A. Alimentary disease in the poor and middle class in London 1773–1815 and in New York poor 1797–1818. Aliment Pharmacol Ther 2002; 16: 1709-1714.
- 12. Spicer CC. The generation method of analysis applied to mortality from tuberculosis. J Hyg (Lond) 1954; 52: 361-368.
- 13. Susser M. Commentary: the longitudinal perspective and cohort analysis. Int J Epidemiol 2001; 30: 684-687.
- 14. Malaty HM, Berenson GS, Wattington WA, et al. Helicobacter pylori acquisition in childhood: 12-year follow-up cohort study in a bi-racial community [abstract]. Gut 1997; 41 Suppl 1: A33.
- 15. Burnet FM. Natural history of infectious disease. 2nd ed. Cambridge: Cambridge University Press, 1953: 209.
- 16. Cullen DJ, Hawkey GM, Greenwood DC, et al. Peptic ulcer bleeding in the elderly: relative roles of Helicobacter pylori and non-steroidal anti-inflammatory drugs. Gut 1997; 41: 459-462.
- 17. Kurata JH, Nogawa AN. Meta-analysis of risk factors for peptic ulcer. Nonsteroidal anti-inflammatory drugs, Helicobacter pylori, and smoking. J Clin Gastroenterol 1997; 24: 2-17.
- 18. Rosenstock S, Jorgensen T, Bonnevie O, et al. Risk factors for peptic ulcer disease: a population based prospective cohort study comprising 2416 Danish adults. Gut 2003; 52: 186-193.
- 19. Lilienfeld AM. Foundations of epidemiology. 1st ed. New York: Oxford University Press, 1976.
- 20. Doll R, Peto R, Wheatley K, et al. Mortality in relation to smoking: 40 years’ observations on male British doctors. BMJ 1994; 309: 901-911.
- 21. History of cigarettes. JTI International. http://www.jti.com/english/tobacco_corner/tobacco_history/cigarette.aspx (accessed Jul 2005).
- 22. Forey B, Hamling J, Lee P, et al. International smoking statistics. A collection of historical data from 30 economically developed nations. 2nd ed. Oxford: Oxford University Press, 2002.
- 23. Doll R, Peto R, Hall E, et al. Mortality in relation to consumption of alcohol: 13 years’ observations on male British doctors. BMJ 1994; 309: 911-918.
- 24. Paffenbarger RS Jr, Wing AL, Hyde RT. Chronic disease in former college students. XIII. Early precursors of peptic ulcer. Am J Epidemiol 1974; 100: 307-315.
- 25. Aldoori WH, Giovannucci EL, Stampfer MJ, et al. A prospective study of alcohol, smoking, caffeine, and the risk of duodenal ulcer in men. Epidemiology 1997; 8: 420-424.
- 26. Katschinski BD, Logan RF, Edmond M, et al. Duodenal ulcer and refined carbohydrate intake: a case-control study assessing dietary fibre and refined sugar intake. Gut 1990; 31: 993-996.
- 27. Sonnenberg A. Dietary salt and gastric ulcer. Gut 1986; 27: 1138-1142.
- 28. Anda RF, Williamson DF, Escobedo LG, et al. Self-perceived stress and the risk of peptic ulcer disease. A longitudinal study of US adults. Arch Intern Med 1992; 152: 829-833.
- 29. Sonnenberg A. Factors which influence the incidence and course of peptic ulcer. Scand J Gastroenterol Suppl 1998; 155: 119-140.
- 30. Raiha I, Kemppainen H, Kaprio J, et al. Lifestyle, stress, and genes in peptic ulcer disease: a nationwide twin cohort study. Arch Intern Med 1998; 158: 698-704.
- 31. Malaty HM, Graham DY, Isaksson I, et al. Are genetic influences on peptic ulcer dependent or independent of genetic factors for Helicobacter pylori infection? Arch Intern Med 2000; 160: 105-109.
- 32. Malaty HM, Engstrand L, Pedersen NL, et al. Helicobacter pylori infection: genetic and environmental influences. A study of twins. Ann Intern Med 1994; 120: 982-986.
- 33. Frost WH. The age selection of mortality from tuberculosis in successive decades. Am J Hyg 1939; 30: 91-96.
Abstract
Helicobacter pylori is established as a cause of peptic ulcer (PU).
Less well recognised is that an epidemic of PU began around the middle of the 19th century, reached a peak at the turn of the century, and is now on the wane.
As the epidemic developed, the risk of PU increased in successive generations throughout life. Then the epidemic diminished in successive generations.
The risk of gastric ulcer (GU) was highest in people born around 1885, while the risk of duodenal ulcer (DU) was highest in those born about 10–30 years later.
H. pylori infection offers an inadequate explanation of the PU epidemic.
Although the epidemic coincided with a major rise in cigarette smoking, PU then declined in spite of an increased incidence of smoking.
None of the other possible causes of ulcer (non-steroidal anti-inflammatory drugs, stress or diet) satisfactorily explains the epidemics of GU and DU and their asynchronicity.
The best, but inadequate, explanation for the epidemic is the coincidence of the acquisition of a new potent strain of H. pylori in childhood and the uptake of smoking in adult life.