




All individuals referred to the clinic are assessed by the current diagnostic criteria (Box 1), with particular attention to the possibility of other conditions that have symptoms and signs in common with Marfan syndrome (Box 2). Box 3 and Box 4 show some of the skeletal and ocular characteristics of Marfan syndrome. Four families are detailed below to demonstrate the potential range of diagnostic dilemmas faced by physicians who consider Marfan syndrome (MFS) as a provisional diagnosis for a patient or family.
The proband presented with lens subluxation and minor skeletal signs of MFS (long limbs). There were no cardiovascular signs. She had normal intelligence. No other family members were affected. Blood levels of homocysteine were found to be elevated at 220 μmol/L (normal range, < 15 μmol/L), and a diagnosis of homocystinuria was made. She responded well to treatment with pyridoxine and betaine. Homocystinuria shares the predisposition of lens subluxation with Marfan syndrome (Box 2), and we have seen a number of other cases at the Marfan clinic. The correct diagnosis ensured that this patient received both appropriate treatment and accurate genetic counselling for her autosomal recessive condition.
The proband was a man in his thirties who had classical features of MFS with lens subluxation, aortic dilatation (requiring surgery) and marked skeletal abnormalities. He had experienced social difficulties as a child and teenager, suffering from bullying and negative body image because of his skeletal deformities. His father appeared normal and his mother had mild chest wall asymmetry. His brother had long limbs, greater than 20° scoliosis, striae and an inguinal hernia. These musculoskeletal features were consistent with MFS, but insufficient to satisfy the diagnostic criteria. The family requested DNA testing to clarify the status of the proband’s brother. DNA from the proband had a splice acceptor site mutation that was absent in his parents and his brother, indicating a spontaneous mutation. Current evidence suggests that spontaneous mutation accounts for at least 25% of MFS cases.2 Risks to family members are different depending on whether the mutation is inherited or spontaneous. For this family, genetic testing confirmed the genetic status of the proband and provided reassurance for other members of the family.
Thirteen members of one sibship and their offspring were examined. The main clinical presentation was lens subluxation with minor skeletal features of MFS. The family was initially said to have familial isolated ectopia lentis (Box 2). However, our examination showed that several children required surgery for mitral valve prolapse, and two adults had aortic dilatation. Linkage to FBN1 was found, indicating that the family was likely to have a mutation in this gene. One adult, whose children had lens subluxation, had few signs of MFS, but was a mutation carrier based on DNA haplotype analysis and family status. This adult is manifesting low expressivity of the mutation. For this family, the confounding variability of phenotype meant that detection of the FBN1 mutation in a young patient had little prognostic value, but did identify individuals who required regular surveillance. This family demonstrates that some carriers of the same FBN1 gene mutation in a family may not fit the diagnostic criteria for Marfan syndrome but nevertheless have a significant risk of aortic dilatation and its consequences. Therefore, echocardiography needs to be performed for all individuals suspected of having MFS.
The proband was a woman in her fifties with dissection of the ascending aorta, aortic arch and abdominal aorta. Her family history was notable for a number of cases of aortic aneurysm or rupture. Some affected individuals had minor skeletal abnormalities, but none had lens subluxation. The family did not meet the diagnostic criteria for MFS and may have an autosomal dominant familial aneurysmal condition. Several genetic loci, including FBN1, have been implicated in familial aneurysms (see Box 2), and one of these may be mutated in the affected individuals. This family illustrates the issues involved in offering DNA testing to families who do not meet the criteria for Marfan syndrome. In such cases, DNA testing is likely to be costly and complex, and may not ultimately result in a molecular diagnosis.
Evaluation at Marfan Clinic routinely involves cardiological assessment including echocardiography, and ophthalmological review including dilatation of the pupils for slit-lamp examination and keratometry. In patients with subluxated lenses, the possibility of homocystinuria is excluded by measurement of plasma homocysteine. Patients are assessed by a clinical geneticist by examination for skeletal and skin features, and the role of DNA testing is discussed. A genetic counsellor records genetic and medical family history and constructs a three-generation pedigree to identify individuals at risk. Because at least 25% of cases of MFS result from spontaneous mutations,2,3 the absence of other confirmed cases in the family does not rule out MFS. The genetic counsellor also provides support for addressing psychosocial issues that may have arisen because of the condition.
It is worth noting that the Marfanoid features which most commonly lead to referral are tall and thin body habitus with hyperextensible joints, but these features have a low specificity and are not included in the major diagnostic criteria. Box 3 shows some of the skeletal features mentioned in Box 1 which are included in the major criterion for the skeletal system.
Normal reference graphs of ascending thoracic aortic diameter related to age and body size are available.4 Individuals with aortic dilatation are referred to a separate clinic for monitoring and ongoing management. Patients with a dilated aorta undergo magnetic resonance angiography. An affected individual’s siblings and offspring with normal cardiac status are reviewed at 1–5-year intervals until age 20. Individuals with aortic diameters at the upper limit of normal are reviewed annually.
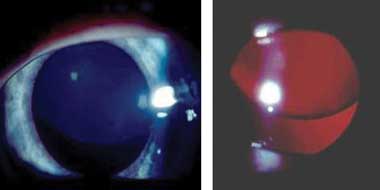
Subluxation of the lens is the major ophthalmic criterion for the diagnosis of Marfan syndrome. The lens is usually displaced superotemporally (Box 4). The zonule fibres are stretched, but still present, resulting in myopic astigmatism. Other minor criteria include iris hypoplasia, displaying a featureless iris, a flat corneal curvature and increased axial length. Other features described previously, including glaucoma, retinal detachment and lens opacity, appear to be a secondary effect of lens subluxation and not a primary effect of Marfan syndrome. Amblyopia occurs in small children as an effect of lens subluxation and resultant refractive error. Lens subluxation may be absent in young children and develop as they age.
At least one individual in 22% of the families seen at the Prince Charles Hospital Marfan Clinic satisfied the international diagnostic criteria for MFS (see Box 11). Additionally, 18% of the families were given other diagnoses, as for Family 1. Box 2 outlines other conditions that share clinical features with MFS. We consider that the 22% diagnostic rate represents an appropriate referral pattern, because of the difficulties in diagnosis and because treatments are available (such as prophylactic β-blockers which may delay aortic dilatation, and pre-emptive surgery which can prevent rupture of an aortic aneurysm, often a fatal event).
As the prevalence of MFS is relatively high (about 1 in 50005), and isolated features of the condition are even more common, many clinicians will encounter potential cases. A diagnosis of MFS raises the possibility of early death from the complications of aortic dilatation and dissection,3 and patients are advised to make lifestyle adjustments to minimise these risks. Pregnancy must be closely monitored in affected women. Patients diagnosed with MFS may find it difficult to obtain life insurance. Misdiagnosis of MFS raises the possibility of inappropriate discrimination by insurance companies and employers, and can lead to improper treatment and surveillance. As outlined above, full cardiovascular, ophthalmological and musculoskeletal evaluation of patients suspected of having Marfan syndrome ensures appropriate diagnosis and circumvents these potential problems.
1 Diagnostic criteria for Marfan syndrome1
2 Genetic conditions which have features in common with Marfan syndrome (MFS)
Increased skin elasticity, fine translucent skin (type IV), bowel rupture (type IV) |
|||||||||||||||
3 Skeletal signs in Marfan syndrome
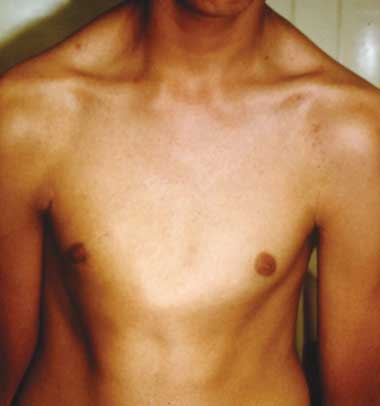 A. Pectus carinatum. 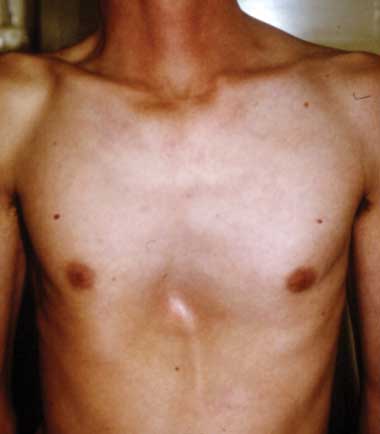 B. Pectus excavatum. This must require surgey to be included in the major criterion (Box 1). 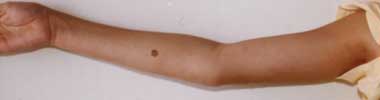 C. Fixed flexion of the elbow. 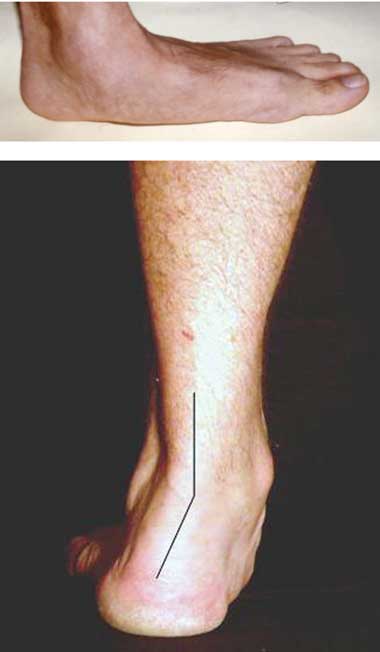 D. Pes planus. This is best diagnosed by examining the foot from behind. A valgus deviation of the hindfoot indicates pes planus. 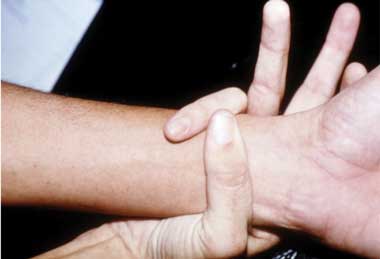 E. Arachnodactyly the wrist sign. 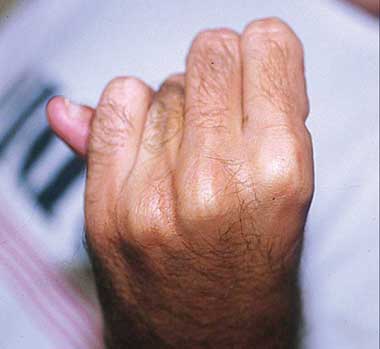 F. Arachnodactyly the thumb sign. |
- Kim M Summers1
- Jennifer A West2
- Madelyn M Peterson3
- Denis Stark4
- James J McGill5
- Malcolm J West2
- 1 School of Molecular and Microbial Sciences, The University of Queensland, Brisbane, QLD.
- 2 The University of Queensland, Prince Charles Hospital, Brisbane, QLD.
- 3 Griffith University, Brisbane, QLD.
- 4 Mater Children's Hospital, Brisbane, QLD.
- 5 Royal Children's Hospital, Brisbane, QLD.
None identified.
- 1. De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996; 62: 417-426.
- 2. Collod-Béroud G, Le Bourdelles S, Adès L, et al. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat 2003; 22: 199-208.
- 3. Pyeritz RE. Marfan syndrome and other disorders of fibrillin. In: Rimoin DL, Connor JM, Pyeritz RE, editors. Emery and Rimoin’s principles and practice of medical genetics. 3rd ed. New York: Churchill Livingstone, 1996: 1027-1066.
- 4. Roman MJ, Devereux RB, Kramer-Fox R, O’Loughlin J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol 1989; 64: 507-512.
- 5. Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet C Semin Med Genet 2005; 139: 4-9.
Abstract
Marfan syndrome (MFS) is a multisystem disorder of connective tissue that is inherited in an autosomal dominant fashion, and results from mutations in the FBN1 gene on chromosome 15.
Diagnosis is challenging as it requires definition of diverse clinical features and input from a variety of specialists.
Genetic testing of FBN1 is time consuming, expensive and complex, and may not solve the diagnostic dilemma.
Failure to make a diagnosis or making an inappropriate diagnosis of MFS has social, lifestyle and medical consequences for the individual as well as the family.