





Volume 182 - Issue 4
Artemisinin-based combination therapies for uncomplicated malaria
Authors: Timothy M E Davis, Harin A Karunajeewa and Kenneth F Ilett
Med J Aust 2005; 182 (4): 181-185. || doi: 10.5694/j.1326-5377.2005.tb06650.x
Published online: 21 February 2005
Published online: 21 February 2005
Abstract
There has been a relentless increase in resistance of malaria parasites to conventional antimalarial drugs, including chloroquine, sulfadoxine–pyrimethamine and mefloquine.
In response to this situation, short-course artemisinin-based combination therapies (ACTs) have been developed.
The World Health Organization has endorsed ACT as first-line treatment where the potentially life-threatening parasite Plasmodium falciparum is the predominant infecting species.
ACTs combine the rapid schizontocidal activity of an artemisinin derivative (artesunate, artemether or dihydroartemisinin) with a longer-half-life partner drug.
Although the use of chloroquine and sulfadoxine–pyrimethamine as partners in ACT improves their efficacy, this may only have value as a short-term measure in patients with a degree of immunity to malaria.
Alternative currently available partner drugs include mefloquine, lumefantrine and piperaquine.
Artesunate–mefloquine is highly effective but is expensive and side effects (mainly neurotoxicity) can be problematic.
Artemether–lumefantrine, the only ACT available in Australia, appears less effective than artesunate–mefloquine and needs to be administered with food to ensure adequate bioavailability.
Dihydroartemisinin–piperaquine is highly effective, well tolerated and relatively inexpensive.
The goal of potent, safe, easy-to-administer and inexpensive ACTs may see trioxolanes in place of artemisinin derivatives, as well as novel partner drugs such as pyronaridine or naphthoquine, in the future.
Malaria remains a major cause of morbidity and death in tropical countries and substantial numbers of Australian travellers are exposed to the risk of malaria each year. Because of the relentless increase in resistance of malaria parasites to conventional drugs, including chloroquine, sulfadoxine–pyrimethamine and mefloquine, new therapeutic approaches have been developed. Prominent among these has been artemisinin-based combination therapy (ACT). This strategy parallels multidrug treatment used successfully in diseases such as HIV and cancer, and combines the rapid schizontocidal effect of an artemisinin compound with a longer-half-life drug. The World Health Organization (WHO) has recently endorsed ACT as the “policy standard” for all malaria infections in areas where Plasmodium falciparum is the predominant infecting species. 1
For a variety of reasons, including cost, ACT is not yet widely available as part of malaria control programs in endemic areas.2 However, there is emerging evidence that ACT is cost-effective, even in sub-Saharan Africa, where there are high transmission rates and meagre economic resources,3 and ACTs are increasingly available for purchase over the counter in tropical countries. In addition, there is now one ACT (artemether–lumefantrine) approved for clinical use in Australia.4 Thus, increasing numbers of Australian tourists and visitors will be treated with ACT. In this article, we review the status of currently available ACTs, with emphasis on the pharmacology of the component drugs and from an Australian perspective.
Artemisinin and its derivatives
The artemisinin group of drugs was discovered in China. A crude extract of the wormwood plant Artemisia annua (qinghao) was first used as an antipyretic 2000 years ago, and its specific effect on the fever of malaria was reported in the 16th century.5 The active constituent of the extract was identified and purified in the 1970s, and named qinghaosu, or artemisinin. Although artemisinin proved effective in clinical trials in the 1980s, a number of semi-synthetic derivatives were developed to improve the drug’s pharmacological properties and antimalarial potency (Box 1).5,6 Several million patients have been treated with these compounds during the past three decades.7
Each artemisinin derivative is highly active against asexual forms of the four species of Plasmodium that infect humans. The initial reduction in parasitaemia is the most rapid of all available antimalarial drugs.6,7 Their half-lives are short (Box 1) relative to the duration of their effect on parasite clearance. This suggests the equivalent of a “post-antibiotic effect”, where there is persistent suppression of bacterial growth following limited exposure to an antimicrobial agent. The artemisinin derivatives are also active against the sexual form of the parasites (gametocytes) taken up by the mosquito and can therefore reduce transmission rates.8 Their exact mechanism of action is unknown. An endoperoxide moiety, which is essential for antimalarial activity, may cause destructive free-radical generation within the parasite and, through the formation of covalent bonds, alter the function of key parasite proteins, including membrane transporters.9,10 Short courses (3–5 days) are associated with recrudescence rates that are typically greater than 25%.6,7 Although such recrudescences can be re-treated successfully with an artemisinin drug, this phenomenon highlights the limitations of monotherapy. There have been no documented cases of parasite resistance to the artemisinin derivatives in humans, and, given the very short time frame of subtherapeutic concentrations, this should continue in the long term.11
The therapeutic index of the artemisinin derivatives is wide. The most common reported adverse effects include nausea, vomiting, bowel disturbance, abdominal pain, headache and dizziness — symptoms that can result from malaria infection itself.12 Mild and reversible haematological and electrocardiographic abnormalities, such as neutropenia and first-degree heart block, are observed infrequently.12 Neurotoxicity, principally in the form of brainstem lesions, was first identified in animals receiving high doses over long periods.13 Neurological side-effects such as ataxia, slurred speech and hearing loss have also been reported in a small numbers of humans,14,15 but are unlikely to be of clinical importance,16,17 especially as there is limited penetration of artemisinin derivatives into the cerebrospinal fluid.18 The pharmacokinetics of artemisinin derivatives such as artesunate are not influenced significantly by severity of infection.19 As vital-organ dysfunction is a feature of severe falciparum malaria, there is no need for dose adjustment in patients with hepatic or renal impairment. There are also no known important drug interactions.20
Of the available derivatives, artesunate has the most favourable pharmacological profile for use in ACT treatment of uncomplicated malaria. The presence of a hemisuccinate group in the molecule confers water solubility and relatively high oral bioavailability. It is rapidly and quantitatively converted in vivo to the potent active metabolite dihydroartemisinin.19 Artemisinin, dihydroartemisinin and artemether are all poorly water-soluble, which means that absorption is slower and less complete.20 Nevertheless, consistent with the wide therapeutic index, the plasma concentrations achieved after oral administration of conventional doses of all of the artemisinin derivatives are substantially greater than those currently required to kill the parasite.20
Antimalarial drug combinations before ACT
Combinations of chemotherapeutic agents can accelerate therapeutic response, improve cure rates and protect the component drugs against resistance. There are several examples of the successful use of antimalarial combinations before the development of ACT, specifically quinine–tetracycline (or quinine–doxycycline), sulfadoxine–pyrimethamine and, most recently, atovaquone–proguanil. With each of these combinations, issues have hampered continued application. Quinine–tetracycline is associated with frequent side effects (including the nausea, dysphoria, tinnitus and deafness of cinchonism), has to be given over a long period (7–10 days) which can affect compliance, and its efficacy is failing in some tropical countries. For sulfadoxine–pyrimethamine, high-grade resistance is increasingly encountered. Atovaquone–proguanil is given as a short-course (3-dose) regimen and is one of the most expensive antimalarial therapies. Despite its relatively recent introduction and limited availability, highly resistant cases have already been reported.21
Candidate partner drugs in ACT
The suggested characteristics of the “ideal” antimalarial combination are shown in Box 2.22 In the case of ACT, the artemisinin component provides a well-tolerated drug with a unique mode of action that clears asexual forms quickly and has gametocidal activity. The partner drug should be one that is also well tolerated and non-toxic, but which is present in the blood at therapeutic concentrations for at least several times the duration of the parasite lifecycle (48 hours in the case of P. falciparum). The characteristics of candidate partner drugs are summarised in Box 3.
Although chloroquine and sulfadoxine–pyrimethamine are readily available and inexpensive (less than US25 cents per treatment course), P. falciparum parasites are resistant to these therapies in most parts of the tropics. Where semi-immunity to malaria is present, the addition of an artemisinin drug such as artesunate can improve efficacy (E1; based on NHMRC levels of evidence23),24 but ACT effectiveness is compromised by using failing drugs as partners.24 Despite this situation, WHO has prioritised artesunate plus amo-diaquine (a chloroquine-like drug which is not available in Australia) and artesunate plus sulfadoxine–pyrimethamine as the second-line and third-line ACT combinations, respectively. 1 Interestingly, where a substantial reduction in the use of chloroquine and sulfadoxine–pyrimethamine has been associated with a reciprocal increase in local parasite sensitivity, the reintroduction of these drugs as part of ACT does not restore efficacy in patients with no immunity to malaria (E3).25
In South-East Asian countries such as Thailand and Cambodia, artesunate–mefloquine has been used quite widely for many years and remains highly effective for uncomplicated falciparum malaria. Under the WHO system of efficacy assessment, more than 95% of patients will remain free of malaria 28 days after such treatment (E2).26 However, the neuropsychiatric effects of mefloquine as well as its relative expense are problematic. No coformulation is available, but in Cambodia there is a blister pack containing tablets of each constituent drug grouped by the doses to be administered over 3 days. It is therefore possible for patients to identify and take the artesunate while leaving the less well tolerated mefloquine.27 This increases the risk of recrudescence and wastes the opportunity to exploit the mutual protection against parasite resistance that has been documented when this combination is used in areas with relatively low-level transmission (E3).28
Artemether–lumefantrine has been given priority as the first-line ACT combination recommended by WHO for uncomplicated malaria. 1 The reasons for this recommendation are unclear. Firstly, artesunate represents a pharmacologically better artemisinin drug than artemether. Secondly, the partner drug, lumefantrine, has to be given with food (preferably containing fat) to ensure adequate absorption and thus efficacy. This bioavailability problem afflicted the chemically related compound halofantrine (which has now been withdrawn because of cardiotoxicity).29 Lumefantrine exhibits no significant cardiotoxicity, but initial studies with a 4-dose artemether–lumefantrine regimen showed a relatively high rate of late treatment failure (E1),30 consistent with the difficulty in ensuring that a febrile, nauseated patient with acute falciparum malaria takes milk or biscuits with each dose to improve bioavailability. A 3-day, 6-dose regimen is now recommended by the manufacturer, but there are insufficient data to confirm that this longer course is as effective as other types of ACT (E1).30 In one study in an area with high-grade mefloquine resistance, the 6-dose artemether–lumefantrine regimen was associated with a 28% treatment failure rate at 14 days (E3).31
Perhaps the most promising form of ACT is dihydroartemisinin–piperaquine. Piperaquine is a bisaminoquinoline frequently used in China in the 1970s and 1980s, when chloroquine resistance became a problem in the south of the country.32 It was rediscovered in the 1990s as a candidate for ACT, and is currently partnered with dihydroartemisinin. There have been two recent Indochinese studies that have demonstrated its effectiveness, with 28-day cure rates greater than 95% (E2).26,33 The 2-day, 4-dose recommended treatment regimen is well tolerated and there are no significant cardiovascular or metabolic side-effects.34 Although the long half-life of piperaquine may theoretically allow resistant parasites to be selected out by the subtherapeutic concentrations present well after treatment,35 this has not been of concern in areas of Thailand where the transmission rate is low and where mefloquine (a drug with a similarly long elimination phase) has been used extensively with artesunate.28
Use of ACT in special groups
Falciparum malaria can be particularly severe in pregnancy. Because of the potential benefits of the artemisinin derivatives in this situation, WHO has published a position statement based on a review of available safety data.36 Use of artemisinin derivatives in 731 pregnancies was not associated with human fetal toxicity, but animal studies have shown significant adverse effects. Because of these latter observations and the limited human data, WHO currently advises against the use of artemisinin drugs in the first trimester, unless in a lifesaving situation where no other drugs are suitable, while, in later pregnancy, alternative therapies should be used if available. There are few data on ACT use in pregnancy.36 If ACT is considered preferable to monotherapy with an artemisinin drug, the partner drug should obviously be chosen with the pregnancy in mind.37 ACT has been used safely in children as young as 1 month4 and paediatric formulations (such as dihydroartemisinin–piperaquine granules32) are available in some cases.
Summary and implications for Australian travellers
In many South-East Asian countries, ACT is now established as first-line treatment for uncomplicated malaria, and it is likely that its use will extend throughout the tropics in future. All currently available artemisinin derivatives are well tolerated, safe and effective, but artesunate has the most favourable pharmacological profile for use in ACT. The choice of partner drug depends on a variety of factors, including parasite resistance patterns, elimination half-life and cost. Two fixed-dose ACT coformulations (artemether–lumefantrine and dihydroartemisinin–piperaquine) are increasingly available in the tropics. The latter may have advantages in terms of ease of administration and effectiveness. Its cost (about US$1 per adult treatment course) is significantly less than the WHO-negotiated discounted price for artemether–lumefantrine (US$2.40)26. However, part of this can be explained by the fact that, unlike artemether–lumefantrine, dihydroartemisinin–piperaquine is not yet manufactured to Good Manufacturing Practice (GMP) standards.
Increasing numbers of Australians will purchase ACT in the tropics, either as presumptive treatment for symptoms suggestive of malaria or as standby therapy. As fake artemisinin derivatives are being sold in pharmacies and shops in South-East Asia,38 it is likely that ACT will follow suit — Australian travellers should be made aware of this disturbing development. The only ACT currently available in Australia is artemether–lumefantrine, which is marketed as Riamet (Novartis). Important messages for patients are given in Box 4. Although there may be better forms of ACT available elsewhere, it is still a very useful addition to the range of antimalarial therapeutic drugs in this country. There are few studies of its efficacy in non-immune travellers,30 but no treatment failures in this context have been reported. Nevertheless, Australian patients taking artemether–lumefantrine should be made aware of the need for full compliance with the instructions, specifically taking the full course and taking each dose with food. A treatment course costs about A$73, compared with A$53 for the atovaquone–proguanil coformulation Malarone (GlaxoSmithKline).
The future for ACT looks promising. Novel chloroquine-like partner drugs, including pyronaridine and naphthoquine, are being assessed and preliminary clinical data are encouraging.39 The combination of artesunate with chlorproguanil–dapsone, two “old” antimalarial drugs, is being developed,40 although haematological toxicity is of concern.41 Although expensive, the combination of atovaquone–proguanil and artesunate proved effective in an area with multidrug-resistant P. falciparum.42 Synthetic trioxolanes43 might prove more cost-effective than currently available artemisinin derivatives and displace them from ACT. Future development of ACT should include continued safety evaluation, especially in pregnant women and young children. The challenge will be to develop a range of new antimalarial combinations that are potent, safe, easy to administer and inexpensive so that the emerging drug-resistant malaria “disaster”44 can be averted.
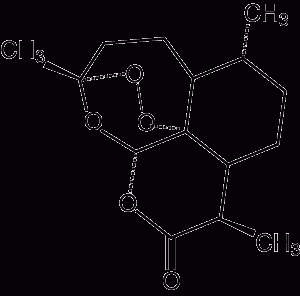
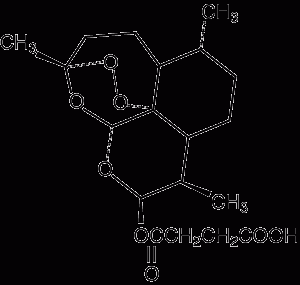
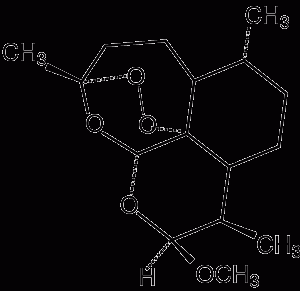
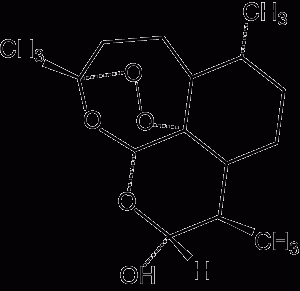
1 Pharmacology of available artemisinin derivatives
Artemisinin Route of administration: Oral, rectal Elimination half-life: 2–3 hours |
|||||||||||||||
Artesunate (artesunic acid) Route of administration: Oral, rectal, intravenous, intramuscular Elimination half-life: 2–5 minutes (converted to dihydroartemisinin) |
|||||||||||||||
Artemether* Route of administration: Oral, intramuscular Elimination half-life: 3–7 hours (converted to dihydroartemisinin) |
|||||||||||||||
Dihydroartemisinin Route of administration: Oral, rectal Elimination half-life: 40–60 minutes |
|||||||||||||||
* Arteether is a closely related compound with an ethyl ether instead of a methyl ether group that has been developed for intramuscular use (elimination half-life, 12–30 hours) |
|||||||||||||||
2 The “ideal” antimalarial combination
Components have different modes of action
No interactions
Short-course regimens (3 days at most)
At least one drug which clears asexual forms rapidly
At least one drug with long half-life (> 4 days)
Well tolerated, low toxicity
Broad spectrum of action (including against gametocytes)
Coformulation possible
Inexpensive
3 Pharmacology of available partner drugs for artemisinin-based combination therapy
Drug |
Elimination half-life |
Coformulation |
Efficacy |
Advantages/disadvantages |
|||||||||||
Chloroquine |
3–14 days |
No |
Depends on degree of resistance, immune status |
Inexpensive. Widely available |
|||||||||||
Fansidar* |
6–9 days for sulfadoxine; 3–4 days for pyrimethamine |
No |
Depends on degree of resistance, immune status |
Inexpensive. Widely available |
|||||||||||
Mefloquine |
2–3.5 weeks |
No |
High |
Neurological/other side-effects. Expensive |
|||||||||||
Malarone† |
12–15 hours for proguanil; 2–3 days for atovaquone |
No |
Few published data |
Expensive. Limited availability |
|||||||||||
Lumefantrine |
4–6 days |
Artemether |
High |
Six-dose regimen. Needs to be given with fat |
|||||||||||
Piperaquine‡ |
3–4 weeks |
Dihydroartemisinin |
High |
Inexpensive. Increasingly available |
|||||||||||
* Roche. †GlaxoSmithKline. ‡Not available in Australia. |
|||||||||||||||
4 Important messages for patients
Artemisinin-based combination therapy (ACT) is now established as first-line treatment for uncomplicated malaria in many South-East Asian countries.
The only ACT available in Australia is artemether–lumefantrine (Riamet).
It is most important to follow the instructions when taking this antimalarial medication — take the whole course and take each dose with food.
Because fake antimalarial therapies are being sold in pharmacies and shops in South-East Asia, it is better to plan ahead and obtain medications for prevention and/or treatment of malaria in Australia before departing.
Competing interests
None identified.
References
- Roll Back Malaria. The RBM partnership’s global response; a programmatic strategy 2004–2008. Available at: http://rbm.who.int/partnership/board/meetings/docs/strategy_rev.pdf (accessed November 2004).
- Attaran A, Barnes KI, Curtis C, et al. WHO, the Global Fund, and medical malpractice in malaria treatment. Lancet 2004; 363: 237-240. i1086024
- Coleman PG, Morel C, Shillcutt S, et al. A threshold analysis of the cost-effectiveness of artemisinin-based combination therapies in sub-saharan Africa. Am J Trop Med Hyg 2004; 71(Suppl 2): 196-204. i1086026
- Australian medicines handbook. Richmond, South Australia: Hyde Park Press, 2004: 215-216. i1086028
- Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. Science 1985; 228: 1049-1055. i1086030
- Hien TT, White NJ. Qinghaosu. Lancet 1993; 341: 603-608. i1086032
- De Vries PJ, Dien TK. Clinical pharmacology and therapeutic potential of artemisinin and its derivatives in the treatment of malaria. Drugs 1996; 52: 818-836. i1086034
- Chen PQ, Li GQ, Guo XB, et al. The infectivity of gametocytes of Plasmodium falciparum from patients treated with artemisinin. Chin Med J 1994; 107: 709-711. i1086036
- Meshnick SR. Artemisinin antimalarials: mechanisms of action and resistance. Med Trop 1998; 58(Suppl): 13-17. i1086038
- Eckstein-Ludwig U, Webb RJ, Van Goethem ID, et al. Artemisinins target the SERCA of Plasmodium falciparum. Nature 2003; 424: 957-961. i1086040
- White NJ. Antimalarial drug resistance. J Clin Invest 2004; 113: 1084-1092. i1086042
- Price R, van Vugt M, Phaipun L, et al. Adverse effects in patients with acute falciparum malaria treated with artemisinin derivatives. Am J Trop Med Hyg 1999; 60: 547-555. i1086044
- Brewer TG, Grate SJ, Peggins JO, et al. Fatal neurotoxicity of arteether and artemether. Am J Trop Med Hyg 1994; 51: 251-259. i1086046
- Miller LG, Panosian CB. Ataxia and slurred speech after artesunate treatment for falciparum malaria. N Engl J Med 1997; 336: 1328. i1086048
- Toovey S, Jamieson A. Audiometric changes associated with the treatment of uncomplicated falciparum malaria with co-artemether. Trans R Soc Trop Med Hyg 2004; 98: 261-267. i1086050
- Kissinger E, Hien TT, Hung NT, et al. Clinical and neurophysiological study of the effects of multiple doses of artemisinin on brain-stem function in Vietnamese patients. Am J Trop Med Hyg 2000; 63: 48-65. i1086052
- Davis TME. Safety evaluations of drugs containing artemisinin derivatives for the treatment of malaria. Clin Infect Dis 2003; 36: 1627-1628. i1086054
- Davis TME, Binh TQ, Ilett KF, et al. Penetration of dihydroartemisinin into cerebrospinal fluid after administration of intravenous artesunate in severe falciparum malaria. Antimicrob Agents Chemother 2003; 47: 368-370. i1086056
- Davis TME, Batty KT, Ilett KF, et al. Pharmacokinetics and pharmacodynamics of intravenous artesunate in severe falciparum malaria. Antimicrob Agents Chemother 2001; 45: 181-186. i1086058
- Ilett KF, Batty KT. Artemisinin and its derivatives. In: Yu VL, Edwards G, McKinnon PS, Peloquin C, editors. Antimicrobial therapy and vaccines. Vol II. Antimicrobial drugs. London: ESun Technologies LLC, 2004: 957-978. i1086060
- Wichmann O, Muehlen M, Gruss H, et al. Malarone treatment failure not associated with previously described mutations in the cytochrome b gene. Malaria Journal 2004; 3: 14. i1086062
- Nosten F, Brasseur P. Combination therapy for malaria: the way forward?. Drugs 2002; 62: 1315-1329. i1086064
- National Health and Medical Research Council. Guide to the development implementation and evaluation of clinical practice guidelines. Canberra: NHMRC, 1999. Available at: www.health.gov.au/nhmrc/publications/pdf/cp30.pdf (accessed Jan 2005). i1086066
- Adjuik M, Babiker A, Garner P, et al: International Artemisinin Study Group. Artesunate combinations for treatment of malaria: meta-analysis. Lancet 2004; 363: 9-17. i1086068
- Nguyen MH, Davis TM, Cox-Singh J, et al. Treatment of uncomplicated falciparum malaria in southern Vietnam: can chloroquine or sulfadoxine-pyrimethamine be reintroduced in combination with artesunate? Clin Infect Dis 2003; 37: 1461-1466. i1086070
- Hien TT, Dolecek C, Mai PP, et al. Dihydroartemisinin-piperaquine against multidrug-resistant Plasmodium falciparum malaria in Vietnam: randomised clinical trial. Lancet 2004; 363: 18-22. i1086072
- Shwe T, Lwin M, Aung S. Influence of blister packaging on the efficacy of artesunate + mefloquine over artesunate alone in community-based treatment of non-severe falciparum malaria in Myanmar. Bull WHO 1998; 76(Suppl 1): 35-41. i1086074
- Nosten F, van Vugt M, Price R, et al. Effects of artesunate-mefloquine combination on incidence of Plasmodium falciparum malaria and mefloquine resistance in western Thailand: a prospective study. Lancet 2000; 356: 297-302. i1086076
- Taylor WR, White NJ. Antimalarial drug toxicity: a review. Drug Safety 2004; 27: 25-61. i1086078
- Omari AA, Gamble C, Garner P. Artemether-lumefantrine for uncomplicated malaria: a systematic review. Trop Med Int Health 2004; 9: 192-199. i1086080
- Poravuth Y, Socheat D, Fandeur T, et al. Efficacy and safety of six dose regimen of Coartem® in the treatment of acute uncomplicated falciparum malaria in Cambodia 2002. In: Gay F, editor. Proceedings of Mekong Malaria Symposium, December 10–13, 2002, Siem Reap, Angkor Wat, Kingdom of Cambodia. (Mekong Malaria Forum, RMCP-EC): 54. i1086082
- Davis TME, Hung TY, Sim IK, et al. Piperaquine: A resurgent antimalarial drug. Drugs 2005; 65: 75-87. i1086084
- Denis MB, Davis TME, Hewitt S, et al. Efficacy and safety of dihydroartemisinin-piperaquine (Artekin) in Cambodian children and adults with uncomplicated falciparum malaria. Clin Infect Dis 2002; 35: 1469-1476. i1086086
- Karunajeewa H, Lim C, Hung T, et al. Safety evaluation of fixed combination piperaquine plus dihydroartemisinin (Artekin®) in Cambodian children and adults with malaria. Br J Clin Pharmacol 2004; 57: 93-99. i1086088
- Hung T-Y, Davis TME, Ilett KF, et al. Population pharmacokinetics of piperaquine in adults and children with uncomplicated falciparum or vivax malaria. Br J Clin Pharmacol 2004; 57: 253-262. i1086090
- World Health Organization. Assessment of the safety of artemisinins in pregnancy: Report of two informal consultations convened by WHO in 2002. (WHO/CDS/MAL/2003.1094, WHO/RBM/TDR/Artemisinin/03.1). Geneva: WHO, 2003: 1-14. i1086092
- Phillips-Howard PA, Wood D. The safety of antimalarial drugs in pregnancy. Drug Safety 1996; 14: 131-145. i1086094
- Newton P, Proux S, Green M, et al. Fake artesunate in southeast Asia. Lancet 2000; 357: 1948-1950. i1086096
- World Health Organization. Antimalarial drug combination therapy. Report of a WHO Technical Consultation. (WHO/CDS/RBM/2001.35). Geneva: WHO, April 4-5, 2001. i1086098
- Development project: Dihydroartemisinin (DHA)-piperaquine (Artekin®). Medicines for Malaria Venture (MMV). Available at: http://www.mmv.org/pages/content_frame.asp?edition=¢er_page=page1_00030004_10.htm&SiteLanguage=1&Nav=00030004 (accessed November 2004).
- Alloueche A, Bailey W, Barton S, et al. Comparison of chlorproguanil–dapsone with sulfadoxine–pyrimethamine for the treatment of uncomplicated falciparum malaria in young African children: double-blind randomised controlled trial. Lancet 2004; 363: 1843-1848. i1086102
- van Vugt M, Leonardi E, Phaipun L, et al. Treatment of uncomplicated multidrug-resistant falciparum malaria with artesunate–atovaquone–proguanil. Clin Infect Dis 2002; 35: 1498-1504. i1086104
- Vennerstrom JL, Arbe-Barnes S, Brun R, et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004; 430: 900-904. i1086106
- Hastings IM, D’Alessandro U. Modelling a predictable disaster: the rise and spread of drug-resistant malaria. Parasitol Today 2000; 16: 340-347. i1086108