




While pituitary disorders are considered uncommon, pituitary adenomas are found in 10%–25% of unselected autopsy series,1 and magnetic resonance imaging reveals pituitary lesions in about 10% of normal individuals.2 The reasons for this discrepancy are not clear. With the advent of high-resolution cranial imaging, unsuspected and often clinically significant pituitary lesions are commonly discovered during investigation of unrelated conditions.
Pituitary disease may present with one or more of the following:
deficiency of one or more pituitary hormones;
hormone excess (especially of prolactin, growth hormone and adrenocorticotropic hormone [ACTH]);
mass effects of an expanding lesion (eg, headache, bitemporal hemianopia); or
an unexpected finding of computed tomography or magnetic resonance imaging (“incidentaloma”).
The recognition that growth hormone should be replaced in adults with growth-hormone deficiency is an important advance in clinical endocrinology. In addition to its role in growth in childhood and adolescence, growth hormone is important in protein and fat metabolism throughout adult life, maintaining muscle mass and reducing visceral fat accumulation. In adult-onset growth-hormone deficiency, present almost invariably in patients with pituitary or hypothalamic disease causing other pituitary-hormone deficiencies, muscle mass is reduced, and fat mass increased. Growth-hormone deficiency impairs physical fitness and psychological well-being, and diminishes quality of life.3 Metabolic consequences include hyperinsulinaemia, reduced glucose tolerance and raised levels of total and low-density lipoprotein (LDL) cholesterol and triglycerides. Growth-hormone replacement reverses the effects of deficiency and often improves quality of life.4
Patients with suspected growth-hormone deficiency should be referred to experienced endocrine centres for diagnostic testing using the insulin tolerance test.5,6 This test distinguishes growth-hormone deficiency from the reduction in growth-hormone secretion that accompanies normal ageing and obesity. The test is performed under supervision and is contraindicated in patients with seizure disorders. Measurement of serum insulin-like growth factor (IGF-1), the growth-hormone-dependent growth factor, is helpful but insufficient, as the level may be within the normal range in up to 30% of adults with growth-hormone deficiency.
Growth-hormone replacement should be undertaken in conjunction with an endocrinologist.5 Recombinant growth hormone is administered by daily subcutaneous injection in a dose range of 0.5–2 U/day, using a calibrated pen device. Dose-related salt and water retention may result in oedema, carpal tunnel syndrome and myalgia. These symptoms resolve with dose reduction. The dose should be adjusted to ensure that serum IGF-1 levels do not exceed the normal range. Growth hormone was approved for treatment of growth-hormone deficiency in adults in Australia in 2001, but has not been included in the Pharmaceutical Benefits Scheme. The annual cost to the patient is $7000–$10 000.
A serum IGF-1 level which exceeds the age- and sex-appropriate reference range confirms clinically suspected acromegaly. Pulsatile secretion and effects of fasting and exercise make elevated serum growth hormone a less reliable indicator.
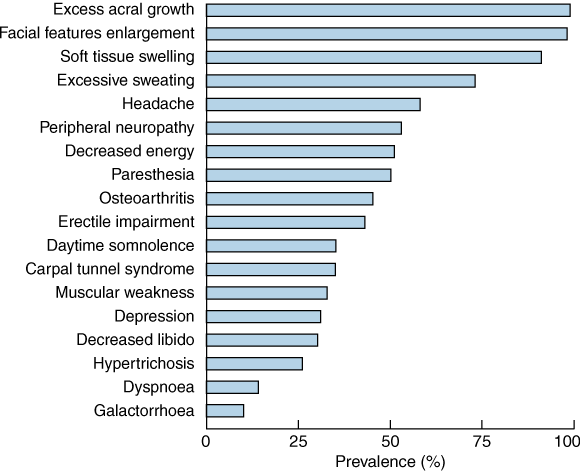
Acromegaly can very rarely be caused by ectopic secretion of growth-hormone-releasing hormone or growth hormone, but over 99% of cases are due to a growth-hormone-secreting pituitary tumour. The incidence of acromegaly is three to four cases per million per year, and the prevalence is 50–70 cases per million, with men and women equally affected. Acromegaly is more aggressive in younger people. The prevalence of clinical features is shown in Box 2. Sleep apnoea is a very common complication which improves with disease control.7 Mortality is increased 1.5 to 3 times in untreated acromegaly, and survival is reduced by 10 years.8 Prognosis is worsened by the presence of hypertension or cardiac disease. Treatment which reduces serum growth hormone level to < 2.5 μg/L, or serum IGF-1 level to the reference range, reduces mortality to that of the general population.9,10
Trans-sphenoidal surgery is regarded as first-line treatment of acromegaly and, in the best centres, normalises growth-hormone secretion in about 70% of patients with a microadenoma (< 1 cm), but in less than 50% of those with a macroadenoma (> 1 cm) because of the difficulty of safely removing all tumour tissue. Pituitary surgery may result in partial or complete hypopituitarism.
Medical therapy may be considered as initial treatment in patients unsuitable for surgery and in those who wish to delay surgery because of potential effects on fertility (see case report, Box 3). Medical therapy has an important role in management of residual growth-hormone excess after pituitary surgery. Radiotherapy may also be used for this purpose, but takes many years to achieve a substantial reduction in growth-hormone secretion, and carries a significant risk of hypopituitarism. Long-acting analogues of somatostatin, the natural inhibitor of growth-hormone secretion, are the drugs of choice,11 and have largely superseded the dopamine agonists bromocriptine and cabergoline. The somatostatin analogues reduce growth-hormone levels in over 90% of patients, normalise IGF-1 level in 50%–60%, and reduce tumour size in most.12 Depot formulations of octreotide and lanreotide provide control for about 28 days and 14 days, respectively. A longer-acting, water-soluble gel formulation of lanreotide, which provides control for 28 days, has recently become available. These analogues often cause gastrointestinal symptoms which are usually mild and self-limiting, and increase the risk of gallstone formation. Symptomatic malabsorption may be helped by pancreatic enzyme supplements.
Pegvisomant, a recombinant growth-hormone analogue which acts as a growth-hormone receptor antagonist, has been shown to be much more effective than somatostatin analogues.13 However, it is unlikely to become the drug of first choice as it does not affect tumour secretion or growth. It is not yet available in Australia.
Most incidentally discovered pituitary masses are small, non-functioning, asymptomatic adenomas. About a third are macroadenomas (> 1 cm), some causing previously unrecognised pituitary hormone deficiency. About 10% secrete hormone, most commonly prolactin. Occasional growth-hormone- and ACTH-secreting adenomas are identified in patients with minimal clinical manifestations.
Serial imaging has shown that most microadenomas do not change in size. 14-16 About 10% of incidental macroadenomas require surgical intervention at the time of diagnosis because of visual field loss or hypopituitarism, and about 25% enlarge significantly, some eventually requiring surgery. However, spontaneous size reduction occurs in about 10%.
Initial management should focus on assessment of tumour size and growth by MRI, delineation of visual fields and detection of hormone excess or deficiency (Box 1). Periodic review by an endocrinologist of patients with asymptomatic, non-functioning presumed macroadenomas is essential to detect enlargement and developing hypopituitarism. Those with visual-field loss should be referred for surgery, usually by the trans-sphenoidal route.
In women, hyperprolactinaemia causes galactorrhoea, and results in menstrual dysfunction and amenorrhoea through inhibition of gonadotropin secretion. In men, it causes secondary hypogonadism and impairs libido and erectile function by a direct, reversible effect on the hypothalamus.
Causes of hyperprolactinaemia are shown in Box 4. In at least 60% of patients with mild hyperprolactinaemia (serum prolactin level up to 3000 mU/L; reference range, < 500 mU/L) that is not caused by medication, imaging shows evidence of a microadenoma (< 1 cm diameter: “microprolactinoma”). Large non-functioning pituitary lesions may also cause mild hyperprolactinaemia by compressing the pituitary stalk, reducing dopamine-mediated inhibition of prolactin secretion. More severe prolactin excess is almost invariably caused by a prolactinoma. Occasional prolactinomas also secrete growth hormone.
In men, most non-drug-induced hyperprolactinaemia is due to a prolactin-secreting macroadenoma or a non-functioning macroadenoma causing pituitary stalk compression, often associated with hypopituitarism and/or compression of the optic chiasm. Postmortem studies reveal a similar prevalence of microprolactinomas in men and women, but these are less often diagnosed in men, perhaps because of the difficulty of detecting subtle effects of mild prolactin excess in men.
Up to 10% of cases of mildly elevated serum prolactin are caused by the presence of circulating antibody–prolactin complexes (“macroprolactinaemia”), which have uncertain physiological significance. Macroprolactinaemia can be detected by most laboratories and should be sought in patients in whom there is uncertainty about the significance of mild prolactin excess.17
Microprolactinoma: Dopamine-agonist therapy (bromocriptine or cabergoline) is the treatment of choice for both micro- and macroprolactinomas. These agents normalise prolactin, restore ovulatory menstrual cycles, and reduce tumour size in at least 90% of cases (see case report, Box 5). In patients with microadenomas, cabergoline (0.5 mg, once or twice weekly) is preferable to bromocriptine (usual dose, 2.5 mg two–three times daily) because of its greater efficacy, better profile of adverse effects and much longer duration of action. 18,19 Dose reduction is often possible after 6–12 months. Long-term observation of microprolactinomas treated with dopamine agonists shows persisting inhibition of tumour growth and apparent resolution in 5%–10%.20 Because serum prolactin is a reliable marker of tumour size, serial imaging is rarely necessary.
Trans-sphenoidal surgery may be considered in the small proportion of patients (< 5%) who are intolerant of dopamine agonists. Major centres report long-term surgical cure of microprolactinomas in about 50% of cases; success rates are likely to be lower in other centres.21 Pituitary surgery may cause permanent hormone deficiency, including hypogonadism and infertility.
Macroprolactinoma: Dopamine-agonist therapy is equally efficacious for treatment of macroprolactinomas and is the initial treatment of choice. In patients with compression of the optic chiasm, substantial tumour shrinkage resulting in recovery of visual field defects often occurs within days of starting therapy18,19 (Box 6). In men, sexual function and testosterone secretion are often restored, and other hormone deficiencies may recover.
Surgery has an important role if visual impairment does not improve during dopamine-agonist therapy, and, in the case of acute haemorrhage causing severe headache, impaired vision and cranial nerve palsies (“pituitary apoplexy”). However, as surgery for macroprolactinomas is both rarely curative and potentially hazardous, dopamine-agonist treatment is the preferred treatment. Radiotherapy may be indicated postoperatively for residual tumours that are resistant to medical therapy.
Pregnancy: Microprolactinomas rarely cause complications during pregnancy. In contrast, 15%–30% of macroprolactinomas enlarge sufficiently to threaten vision. Close monitoring of visual fields is essential, followed by MRI if fields are abnormal. Dopamine agonists are usually effective in shrinking symptomatic tumours, and there is no evidence that these agents increase the risk of fetal malformations. Bromocriptine may be preferred over cabergoline because of greater experience with this drug in pregnancy.20
Most cases (70%–80%) of Cushing’s syndrome are caused by an ACTH-secreting pituitary microadenoma (“Cushing’s disease”). Ectopic ACTH secretion by small-cell lung cancer or other solid tumours causes a distinct, rapidly progressive syndrome of pigmentation, severe myopathy and hypokalaemia which is readily recognised. However, small bronchial carcinoids and other neuroendocrine tumours may cause ACTH-dependent Cushing’s syndrome that is clinically indistinguishable from that caused by a pituitary microadenoma.22,23 ACTH-dependent hypercortisolism may also be caused by depression, illness and alcoholism, the biochemical and clinical features of which may be difficult to distinguish from classical Cushing’s syndrome.23 ACTH-independent causes of Cushing’s syndrome include adrenal adenoma, adrenal carcinoma and bilateral adrenal hyperplasia.
Weight gain, central fat distribution, dorsal fat pad (“buffalo hump”), hirsutism, diabetes and hypertension have low specificity for diagnosis of Cushing’s syndrome. Skin fragility, easy bruising, purple striae, and proximal myopathy are more reliable indicators.23
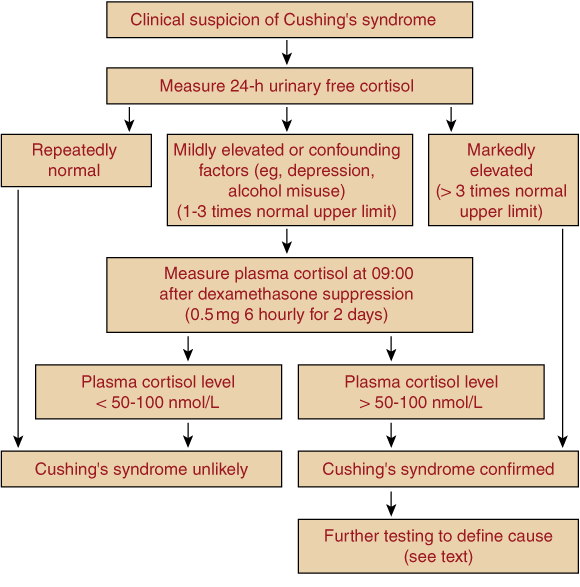
Useful tests for diagnosing glucocorticoid excess are the overnight low-dose (1 mg) dexamethasone suppression test and measurement of 24-hour urine free cortisol23 (Box 7). Although the overnight suppression test has high sensitivity for Cushing’s syndrome, false-positive results are common, especially in depression. Specific, extracted assays of 24-hour urine free cortisol (with reference ranges < 330 nmol/24 h) have high sensitivity (> 95%); the false-positive rate is < 5% in obesity, but higher in depression and alcohol dependence.23 Unextracted and other, less-specific assays of urine free cortisol, which cite upper normal limits as high as 400–900 nmol/24 h, have lower diagnostic value.
In patients with suggestive clinical features and no confounding factors (eg, illness, depression or alcohol dependence), a marked elevation of 24-hour urine free cortisol on several occasions is usually sufficient to diagnose Cushing’s syndrome. When diagnostic uncertainty remains, the patient should be referred for further investigations, including the 2-day low-dose (0.5 mg 6-hourly) dexamethasone suppression test, measuring plasma rather than urine cortisol for greater diagnostic accuracy.23 Suppression of 09.00 plasma cortisol level to less than 50–100 nmol/L makes Cushing’s syndrome unlikely, whereas higher levels are generally confirmatory. Midnight plasma cortisol level also provides excellent discrimination, but requires hospital admission.23
Once endogenous cortisol excess has been confirmed, then plasma ACTH level should be measured to distinguish between ACTH-dependent and ACTH-independent causes. An undetectable ACTH level points to an adrenal source of cortisol, while a measurable or elevated ACTH level indicates pituitary or ectopic ACTH secretion. Further investigations to determine the cause of ACTH-dependent Cushing’s syndrome should be undertaken in an endocrine unit. Suppression of 24-hour urine free cortisol by at least 80% by high-dose dexamethasone (2 mg 6-hourly for 2 days) has traditionally been used to diagnose pituitary-dependent Cushing’s syndrome. However, up to 10% of patients with ectopic ACTH secretion also show dexamethasone suppressibility, and suppression is impaired in at least 10% of patients with pituitary-dependent Cushing’s syndrome. Consequently, the value of the 2-day and overnight high-dose dexamethasone suppression test has been questioned.23
The use of corticotropin-releasing hormone stimulation to distinguish between pituitary and ectopic ACTH secretion has been advocated.24 However, corticotropin-releasing hormone is not readily available in Australia.
Gadolinium-enhanced pituitary MRI should be performed in all patients with ACTH-dependent Cushing’s syndrome. A pituitary microadenoma is visualised in 50%–60% of those eventually proven to have Cushing’s disease by trans-sphenoidal exploration.23 Although MRI shows presumed microadenomas in 10% of normal subjects, the finding of a definite pituitary lesion has high specificity in the context of biochemically confirmed ACTH-dependent Cushing’s syndrome.
Bilateral inferior petrosal sinus sampling for ACTH is the best available method for confirming or excluding pituitary origin of ACTH secretion in Cushing’s syndrome.23 This technique has over 95% diagnostic accuracy in specialised units, but has a small risk of serious complications, and should only be done in experienced centres. Lateralisation of pituitary microadenomas by this technique is unreliable because of highly variable patterns of pituitary venous drainage. Patients whose investigation results raise the possibility of ectopic ACTH secretion should undergo high-resolution CT of the chest.
Selective trans-sphenoidal surgery by an experienced pituitary surgeon is curative in about 80% of patients with an ACTH-secreting pituitary microadenoma. In those with persistent or recurrent disease, options include further pituitary surgery, pituitary irradiation, medical therapy with ketoconazole or other agents, or bilateral adrenalectomy.25
1: Baseline endocrine evaluation of pituitary function*
Serum levels of the following hormones should be measured in a morning blood sample:†
Prolactin
Luteinising hormone, follicle-stimulating hormone and testosterone or oestradiol
Thyroid-stimulating hormone and thyroxine
Adrenocorticotropic hormone and cortisol
Insulin-like growth factor-1
* As serum levels of tropic hormones are often “normal” in patients with hypopituitarism, it is essential that levels of target-gland hormones are also measured (ie, thyroxine, cortisol and testosterone or oestradiol).
† Cortisol and testosterone levels are highest in the morning.
3: Case report — acromegaly
 Presentation: A 32-year-old, recently married
man presented with a 12-month history of arthralgia, hyperhidrosis, headaches, somnolence, and complaints from his wife about his heavy snoring.
Presentation: A 32-year-old, recently married
man presented with a 12-month history of arthralgia, hyperhidrosis, headaches, somnolence, and complaints from his wife about his heavy snoring.
Investigation: A clinical suspicion of acromegaly was confirmed by raised serum levels of growth hormone (45 μg/L; reference range [RR], < 2 μg/L) and insulin-like growth factor (IGF-1) (120 nmol/L; RR, 25–55 nmol/L). Pituitary function was otherwise normal. Magnetic resonance imaging (MRI) showed an enlarged pituitary fossa harbouring a soft tissue mass extending into the suprasellar cistern and abutting the optic chiasm. Ophthalmic examination revealed normal visual acuity and fields.
Management: As surgery might impair fertility and was unlikely to be curative because of the size of the tumour, medical therapy was begun with subcutaneous octreotide (100 mg) three times daily.
Course: Symptoms improved rapidly, and biochemical assessment 3 months later revealed a serum level of growth hormone of 3 μg/L, and serum IGF-1 of 40 nmol/L.
A repeat MRI scan at 12 months showed a significant reduction in tumour size.
The patient’s wife conceived during his second year of treatment. Biochemical control remained excellent, and was maintained when treatment was changed to monthly injections of the slow-release preparation octreotide when this became available.
Somatostatin analogues are effective and well tolerated and allow greater treatment flexibility and timing of surgery and radiotherapy. Interim disease control with these drugs allows patients to defer surgery and radiotherapy, which carry a risk of hypopituitarism, until they have completed their families. These drugs are available under the Pharmaceutical Benefits Scheme for treatment of acromegaly by hospital-affiliated specialists.
IGF-1 (previously called somatomedin) mediates the anabolic effects of growth hormone and is an excellent biochemical measure of disease control.
4: Causes of hyperprolactinaemia
Physiological
Drugs
Pituitary stalk damage
|
Prolactinoma
Hypothalamic lesion
Other
|
||||||||||
5: Case report — microprolactinoma
Presentation: A 28-year-old woman presented with amenorrhoea since she stopped taking the oral contraceptive pill 6 months before, hoping to conceive. She had also noticed that a little milky fluid could be expressed from both breasts. Her menstrual cycle was regular before starting the contraceptive pill. She had no recent weight change, hirsutism or acne. She did not exercise excessively. Her health was otherwise good, and she had normal vision. A recent pregnancy test was negative. Her only medication was paracetamol for longstanding, occasional mild headaches.
Examination: Body mass index was 24 kg/m2 (reference range [RR], 20–25 kg/m2). There was bilateral expressible galactorrhoea, with no other breast abnormality. She was clinically euthyroid, with no goitre. Visual fields were normal. Blood pressure was 110/70 mm Hg. No abnormalities of the heart, chest or abdomen were apparent on examination.
What was the most likely diagnosis?
Her regular menstrual cycle before starting oral contraceptives and lack of recent weight loss or other medical problems made functional hypothalamic amenorrhoea unlikely.
Premature menopause should be excluded by confirming that serum level of follicle-stimulating hormone (FSH) was not elevated, but is also unlikely.
Although there was no clinical evidence of androgen excess, this remained a possibility.
The history and physical findings were typical of mild hyperprolactinaemia caused by a microprolactinoma. Initial effects of prolactin excess were presumably masked by the contraceptive pill. Several studies have shown no increase in the incidence of hyperprolactinaemia with oral contraceptive use.20
Investigations: Serum hormone levels were: FSH, 3 IU/L (RR, 2–8 IU/L); luteinising hormone, 2 IU/L (RR, 2–8 IU/L); oestradiol, 120 pmol/L (RR follicular phase, 120–600 pmol/L); prolactin, 2200 mIU/L (RR, < 500 mIU/L); testosterone, 1.2 nmol/L (1.0–2.8 nmol/L); thyroid-stimulating hormone, 1.1 mU/L (RR, 0.3–4.3 mU/L); thyroxine, 14 pmol/L (RR, 9–19 pmol/L).
These results confirmed hyperprolactinaemia and excluded premature menopause and androgen excess.
Was pituitary imaging necessary? If so, by which method?
This patient’s moderate hyperprolactinaemia was most likely due to a microprolactinoma, but could also be the result of a larger lesion or other abnormality. Consequently, imaging was essential. While magnetic resonance imaging (MRI) is more likely to visualise a microadenoma and clearly defines the relationship of larger pituitary lesions to the optic chiasm and other adjacent structures, computed tomography of the pituitary is adequate to exclude larger lesions and sufficient in most cases of mild to moderate prolactin excess.
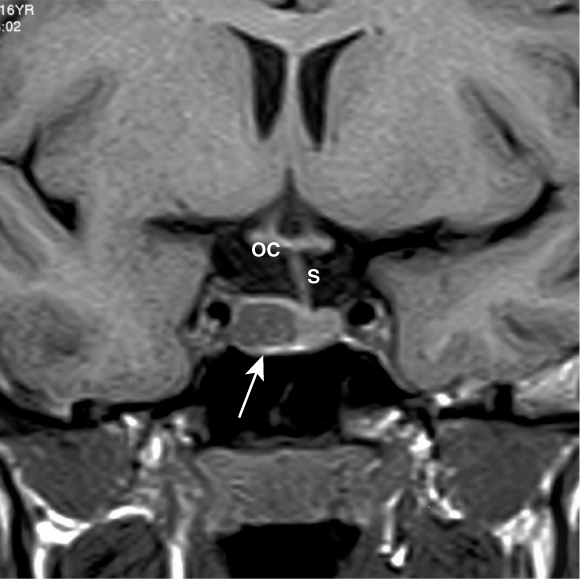
Management: An MRI scan (coronal view above) showed a 1 cm mass of low signal intensity within the right lobe of the pituitary gland (arrow), displacing the pituitary stalk to the left, characteristic of a small prolactinoma. Optic chiasm is labelled OC.
Cabergoline (0.5 mg twice weekly) was begun. The galactorrhoea resolved within 2 weeks, and regular 28-day menstrual cycles resumed. The patient discontinued cabergoline therapy after confirmation of pregnancy 3 months later.
What monitoring is appropriate during pregnancy?
While there is no evidence of adverse effects in pregnancy, dopamine-agonist therapy should be stopped once pregnancy is confirmed. Routine visual field monitoring is not necessary, as the risk of symptomatic enlargement is very low (< 2%). Prolactin levels increase markedly in normal pregnancy, and monitoring of serum prolactin is not helpful. However, visual field asessment and pituitary MRI should be arranged in the event of unusual headache or visual symptoms.20
Course: The pregnancy was uncomplicated. After weaning her baby, the patient returned because of persistent lactation, failure of periods to return, and for contraceptive advice. Serum prolactin level was 2000 mIU/L.
What longer-term management was appropriate?
The current serum prolactin level was similar to the level at presentation, implying that the microprolactinoma had changed little in size. Cabergoline therapy should be resumed to suppress lactation and restore ovarian function, and should be continued long-term to prevent osteoporosis. If necessary, the oral contraceptive pill can be combined with dopamine-agonist therapy.
6: Large prolactinoma before and after cabergoline therapy
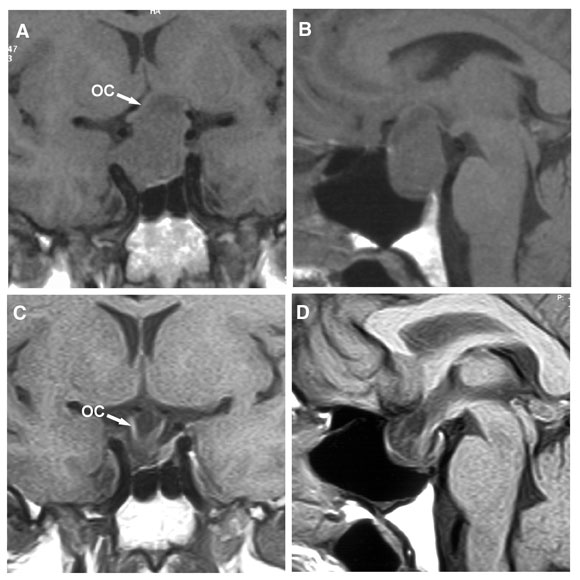
Magnetic resonance imaging in a 47-year-old man who presented with bitemporal visual field loss. (A, B) Before treatment with cabergoline, showing a large mass expanding the pituitary fossa, with substantial suprasellar extension elevating the optic chiasm (OC). (C, D) 6 months after starting cabergoline therapy (0.5 mg twice weekly), showing marked shrinkage of the mass, which is now entirely within the pituitary fossa, and decompression of the optic chiasm, which has descended into the fossa. The patient’s vision improved within a week of starting cabergoline. After 3 weeks, visual fields were almost normal, and serum prolactin level had fallen from 39 000 mU/L to 563 mU/L (reference range, < 330 mU/L).
- 1. Burrow GN, Wortzman G, Rewcastle NB, et al. Microadenomas of the pituitary and abnormal sellar tomograms in an unselected autopsy series. N Engl J Med 1981; 304: 156-158.
- 2. Hall WA, Luciano MG, Doppman JL, et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med 1994; 120: 817-820.
- 3. Cuneo RC, Salomon F, McGauley GA, Sonksen PH. The growth hormone deficiency syndrome. Clin Endocrinol 1992; 37: 387-397.
- 4. Carroll PV, Christ ER, Bengtsson BA, et al. Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. J Clin Endocrinol Metab 1998; 83: 382-395.
- 5. Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of adults with growth hormone deficiency: summary statement of the Growth Hormone Research Society Workshop on Adult Growth Hormone Deficiency. J Clin Endocrinol Metab 1998; 83: 379-381.
- 6. Hoffman DM, O’Sullivan AJ, Baxter RC, Ho KKY. Diagnosis of growth hormone deficiency in adults. Lancet 1994; 343: 1064-1068.
- 7. Grunstein RR, Ho KKY, Sullivan CE. Effect of somatostatin analog (octreotide) on sleep apnea in acromegaly. Ann Intern Med 1994; 121: 478-483.
- 8. Rajasoorya C, Holdaway IM, Wrightson P, et al. Determinants of clinical outcome and survival in acromegaly. Clin Endocrinol 1994; 41: 95-102.
- 9. Giustina A, Barkan A, Casanueva FF, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab 2000; 85: 526-529.
- 10. Orme SM, McNally RI, Cartwright RA, Belchez PE. Mortality and cancer incidence in acromegaly: a retrospective cohort study. J Clin Endocrinol Metab 1998; 83: 2730-2734.
- 11. Lamberts SWJ. Octreotide. N Engl J Med 1996; 334: 246-254.
- 12. Burt MG, Ho KKY. Comparison of efficacy and tolerability of somatostatin analogs and other therapies for acromegaly. Endocrine 2003; 20: 299-305.
- 13. Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med 2000; 342: 1171-1177.
- 14. Feldkamp J, Santen R, Harms E, et al. Incidentally discovered pituitary lesions: high frequency of macroadenomas and hormone secreting adenomas — results of a prospective study. Clin Endocrinol 1999; 51: 109-113.
- 15. Donovan LE, Corenblum B. The natural history of the pituitary incidentaloma. Arch Intern Med 1995; 155: 181-183.
- 16. Reincke M, Allolio B, Saeger W, et al. The incidentaloma of the pituitary gland. Is neurosurgery required? JAMA 1990; 264: 2772-2996.
- 17. Fahle-Wilson MN, Ahlquist JA. Hyperprolactinaemia due to macroprolactins: some progress but still a problem. Clin Endocrinol 2003; 58: 683-685.
- 18. Webster J, Piscitelli G, Polli A, et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. N Engl J Med 1994; 331: 904-909.
- 19. Ferrari CI, Abstract R, Bevan JS, et al. Treatment of macroprolactinoma by cabergoline: a study of 85 patients. Clin Endocrinol 1997; 46: 409-413.
- 20. Molich ME. Medical treatment of prolactinomas. Endocrinol Metab Clin North Am 1999; 28: 143-169.
- 21. Molich ME, Thorner MO, Wilson C. Therapeutic controversy: management of prolactinomas. J Clin Endocrinol Metab 1997; 82: 996-1000.
- 22. Orth DN. Medical progress: Cushing’s syndrome. N Engl J Med 1995; 332: 791-803.
- 23. Newell-Price J, Trainer P, Besser M, Grossman A. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushingoid states. Endocr Rev 1998; 19: 647-672.
- 24. Yanovski JA, Cutler Jr GB, Chrousos GP, Nieman LK. Corticotropin-releasing hormone stimulation following low-dose dexamethasone administration. A new test to distinguish Cushing’s syndrome from pseudo-Cushingoid states. JAMA 1993; 269: 2232-2238.
- 25. Utiger RD. Treatment, and retreatment, of Cushing’s disease. N Engl J Med 1997; 336: 215-217.
Abstract
Pituitary adenomas are found in 10%–25% of unselected autopsy series and are evident in about 10% of asymptomatic individuals by magnetic resonance imaging.
Diagnosis of pituitary disorders is often delayed by lack of awareness and the subtlety of symptoms and signs.
Hypopituitarism is suspected when peripheral hormone concentrations are low without an elevation in the corresponding pituitary tropic hormone(s).
Severe adult-onset growth-hormone deficiency results in reduced muscle mass, increased fat mass and diminished quality of life, which are reversed by growth hormone replacement therapy.
While trans-sphenoidal surgery remains first-line treatment for acromegaly, drug treatment has an important role in controlling residual growth-hormone excess and, in some circumstances, as first-line treatment.
Dopamine-agonist therapy (cabergoline or bromocriptine) is the treatment of choice for micro- and macroprolactinomas.
In patients with suggestive clinical features, elevated 24-hour urine free cortisol level is usually sufficient to diagnose endogenous Cushing’s syndrome; careful additional investigation is needed to determine whether the cause is Cushing’s disease (pituitary adenoma secreting adrenocorticotropic hormone [ACTH]), ectopic ACTH secretion or adrenal disease.