





Volume 176 - Issue 2
Sickle-cell/β+-thalassaemia in a Papua New Guinean: the first reported case of the sickle gene in Papua New Guinea
Authors: Evelyn K Lavu, John D Vince and Gilchrist Oswyn
Med J Aust 2002; 176 (2): 70-71. || doi: 10.5694/j.1326-5377.2002.tb04288.x
Published online: 21 January 2002
Published online: 21 January 2002
Abstract
We document the first case of the sickle-cell gene — Hb-S gene importation leading to Hb-S/β-thalassaemia double heterozygosity — in the apparently previously Hb-S gene-free setting of Papua New Guinea.
The sickle haemoglobin gene has deleterious health effects in people who are homozygous. However, it does confer protection against malaria and has concentrated, with other protective haemoglobinopathies, in areas of the world where malaria is endemic.1 It is found widely in Africa, the Mediterranean, the Middle East and parts of India, and in communities of African descent in countries such as the United States and the Caribbean,2 but has never previously been reported in Papua New Guinea. This seems surprising in a country where malaria is endemic, where the thalassaemias are common,3-5 where there is a wide variety of G6PD variants6,7 and where Southeast Asian ovalocytosis (SAO) is also common. 8,9
Here, we report the first documented case of the sickle-cell gene in a Papua New Guinean.
Clinical record
A four-year-old boy from the Oro Province of Papua New Guinea was admitted to hospital with pain over the left upper abdominal quadrant after a fall. He had been admitted on previous occasions with recurrent anaemia. He was pale, with a temperature of 38°C. His spleen was palpable 6 cm below the left subcostal margin. There were no other significant findings.
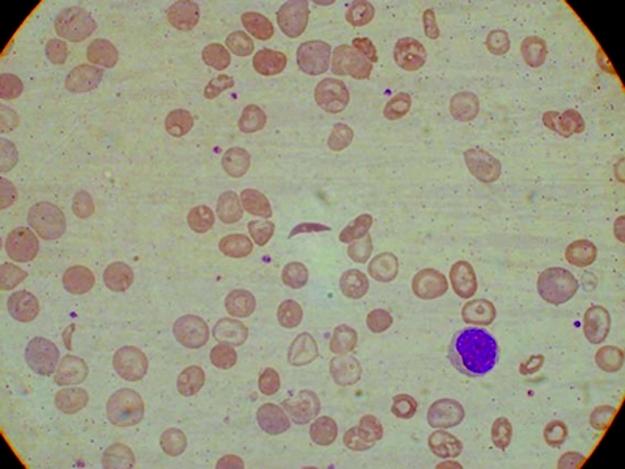
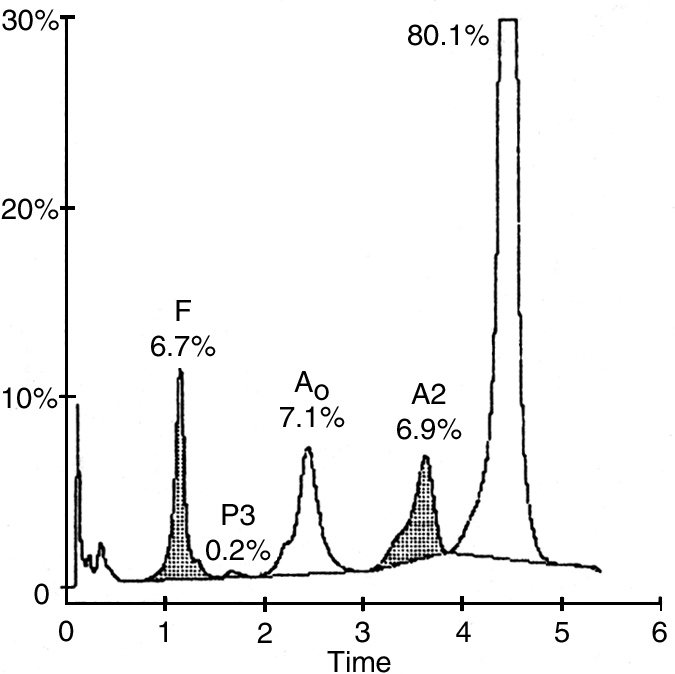
Full blood examination showed a haemoglobin concentration of 82 g/L (reference range, 118–142 g/L), a white cell count of 14.5 x 109/L (reference range, 5–15 x 109/L), with a normal differential count and a platelet count of 610 x 109/L (reference range, 150–400 3 109/L). Red-cell indices were not part of the routine report from the provincial laboratory. The blood film showed sickle cells, hypochromic microcytic cells and many target cells (Box 1). The thick blood film showed Plasmodium falciparum trophozoites at a density of 10 parasites per 100 high power fields. The patient's sickling test was positive. Cation-exchange high-performance liquid chromatography (HPLC) analysis of the haemoglobin (Hb) showed an abnormal peak comprising approximately 80% of the total (Box 2). HbA comprised 7.1% (reference range, > 95%), HbA2 comprised 6.9% (reference range, 1.9%–3.3%) and HbF comprised 6.7% (reference range, 0.0%–1.4%). The patient's haemoglobin electrophoresis strip (on cellulose acetate using a Tris-EDTA boric acid buffer, pH 8.4) showed an abnormal haemoglobin, moving in the position of HbS. This abnormal haemoglobin was confirmed on Titan gel acid electrophoresis. These findings are consistent with double heterozygosity for sickle-cell haemoglobin and β+-thalassaemia trait.10 Electrophoresis of blood from our patient's mother showed abnormal haemoglobin comprising 33.1%, consistent with the heterozygous HbS state (HbAS). Levels of HbA2 and HbF were normal. The father's blood did not contain abnormal haemoglobin, but quantitation showed an HbA2 level of 5.2% and an HbF level of 0.7%, consistent with β-thalassaemia minor. Similar results were obtained for the patient's younger sibling.
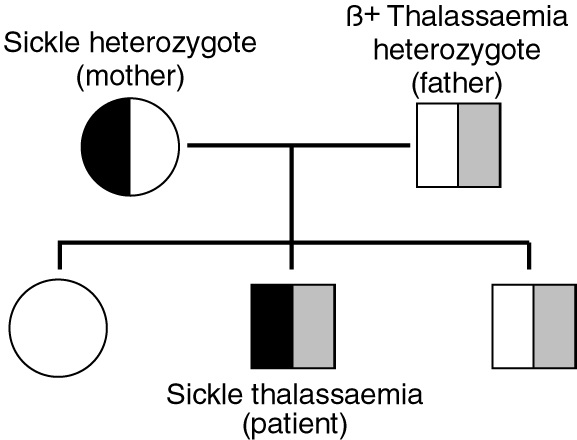
Discussion with the parents after sickle-cell/β+-thalassaemia was confirmed disclosed that the mother's grandmother was either African or of mixed Solomon Islands and African parentage. She had originally come to Papua New Guinea early in the 20th century as a missionary and had married into the local community. The patient's immediate pedigree is shown in Box 3.
Discussion
Marriage outside traditional boundaries is generally accepted as being genetically beneficial. Our case report indicates the rare corollary of such wisdom, in which a gene has been introduced into the community which, while beneficial in normal heterozygotes, is deleterious in homozygotes and in combination with other abnormal genes. The fact that the gene is not clinically expressed in the heterozygous state has disguised its presence until manifestation in our patient with the serious clinical effects resulting from double heterozygosity. It is highly probable that the gene is established in other Papua New Guinean descendants of our patient's great-grandmother.
It is almost inevitable that patients with double heterozygosity and patients with homozygous Hb-S disease will pose additional problems of management and counselling in the future. Patients with sickle-cell/β+-thalassaemia present similarly to those with sickle-cell anaemia and should be managed in the same way.
Acknowledgements
We gratefully acknowledge the patient's family for giving permission to report the case, and the help of the medical laboratory technicians of Popondetta General Hospital. We thank Dr Max Steward of Cairns Pathology and the staff of Sullivan Nicolaides Pathology, Brisbane, for their assistance with the sickling test and with HPLC and electrophoretic analyses. We are grateful to the staff of the Audio Visual Unit of the Medical Library, School of Medicine and Health Sciences, UPNG, for help with preparation of the figures.
References
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998; 11: 1-51. pgfId-1063070
- Weatherall DJ, Provan AB. Red cells I: inherited anaemias. Lancet 2000; 355: 1169-1175. CACGHFCI
- Yenchitsomanus P, Summers KM, Board PG, et al. Alpha thalassaemia in Papua New Guinea. Hum Genet 1986; 74: 432-437. CACBBEAC
- Smiley M. Beta thalassaemia in Papua New Guinea. Ann Trop Paediatr 1986; 6: 175-177. pgfId-1061072
- Allen SJ, O'Donnell A, Alexander NDE, et al. β+Thalassaemia protects children against disease caused by other infections as well as malaria. Proc Natl Acad Sci USA 1997; 94: 14736-14741. CHDJGJHB
- Nurse GT. Haematological genetics. Part 2: Oceania. Clin Haematol 1981; 10: 1051-1067. i1061074
- Chockkalingam K, Board PG, Nurse GT. Glucose-6-phosphate dehydrogenase deficiency in Papua New Guinea. The description of 13 new variants. Hum Genet 1982; 60: 189-192. i1061076
- Amato D, Booth PB. Hereditary ovalocytosis in Melanesians. PNG Med J 1977; 20: 26-32. i1061078
- Sergeantson S, Bryson K, Amato D, Babona D. Malaria and hereditary ovalocytosis. Hum Genet 1977; 37: 161-167. i1061080
- White JM, Marsh GW. Investigation of the abnormal haemoglobins and thalassaemias. In: Dacie JV, Lewis SM, editors. Practical haematology. 7th ed. London: Churchill Livingstone, 1991: 227-266. i1061082