





Legalising mitochondrial donation will present a promising path forward to reduce mitochondrial disease transmission in affected families
Australia is poised to make historic legislative changes that, for the first time, would enable couples affected by maternally inherited mitochondrial disease the opportunity of having unaffected genetically related children.
Mitochondrial diseases encompass a broad range of multi‐organ disorders, ranging from mild to life‐threatening conditions that manifest across all age groups. No cures are available, only symptomatic treatments.1 These heritable diseases are caused by the dysfunction of mitochondria — organelles that produce essential energy (adenosine triphosphate [ATP]) to power all crucial cellular functions. Energy‐demanding tissues, such as brain and muscle, are most commonly, but not exclusively, affected. Mitochondrial disorders are caused by mutations in either the mitochondria’s own DNA (mtDNA; many copies of a 16 569 base pair circular genome) or in the 3.2 billion base pair nuclear DNA (nDNA; in 46 chromosomes) present in each cell.2 Unlike nDNA, which is inherited from both parents, mtDNA is maternally inherited, and mtDNA variants cause maternally inherited mitochondrial disease (Box 1). The transmission of mutant mtDNA into individual oocytes, and subsequent distribution to different embryonic cells after fertilisation, is a complex and largely unpredictable process that continues throughout life. Most affected individuals have both normal and mutant mtDNA in each cell (known as heteroplasmy). Variable mutational loads in different individuals and different cell types result from unequal segregation of normal and mutant mtDNA as cells divide.2 Programmed reduction in mitochondrial copy number in primordial germ cells can select against particular mtDNA variants, causing a shift in heteroplasmy from one generation to the next.3,4 The cellular dysfunction caused by mtDNA mutations depends on the cell’s specific energy requirements as well as the total amount and ratio of normal to mutant mtDNA. Substantial evidence suggests that the higher the mutant mtDNA load, the higher the risk of severe disease, although there are many discrepancies.5 Over 250 pathological mtDNA mutations have been identified,1 with each likely to have their own threshold load for causing mitochondrial dysfunction.6,7 This complex and nuanced genetic transmission puts mtDNA‐mediated mitochondrial disease in a separate category of inherited genetic diseases to nDNA mutations, which are transmitted by largely predictable Mendelian genetics. Patients with mitochondrial disease require diagnosis and familial risk evaluation by highly skilled clinicians who understand this complexity.
In Australia, the incidence of mtDNA mutations is predicted to be at least 1:250, with several hundred families already diagnosed, although many carriers remain unidentified.4,8 Some families have multiple generations of affected individuals, often with devastating consequences. Their health care needs present enormous emotional, physical, social and financial burdens on families, leading many couples to seek options to prevent disease transmission to their offspring. Their current choices include voluntary childlessness, adoption, using eggs donated by unaffected women, or prenatal and pre‐implantation genetic diagnosis. For couples wanting to have genetically related children, prenatal and pre‐implantation genetic diagnosis are not reassuring options because all of the woman’s oocytes may produce embryos with high levels of mutant mtDNA in some cells.9 Recent developments in mitochondrial donation now present a promising path forward to reduce disease transmission in these families by replacing faulty mitochondria containing mutant mtDNA with healthy mitochondria containing normal mtDNA.10 To ensure all cells of the offspring contain healthy mitochondria, this replacement must be done at the one‐cell stage of conception, using procedures to manipulate mature or newly fertilised oocytes that are currently prohibited under Australian legislation. After lengthy and extensive scientific review and public consultation, the United Kingdom changed its legislation in 2015 to allow mitochondrial donation under specified regulation.11 Following this lead, the Australian Senate initiated an inquiry in 2018 to consider the appropriateness of the technology in the Australian context. A Citizens’ Jury and a National Health and Medical Research Council (NHMRC) Mitochondrial Expert Working Committee and Citizens’ Panel facilitated wide‐ranging community consultation12,13 over 3 years that preceded drafting of the Mitochondrial Donation Law Reform (Maeve’s Law) Bill 2021, currently under Parliamentary review. In this article, we outline the scientific and ethical issues raised by mitochondrial donation and the changes to legislation needed before its implementation in Australia.
The technology and the law
Mitochondrial donation refers to assisted reproductive technologies used to uncouple the inheritance of an affected woman’s nDNA (normal) from her mtDNA (mutant). Two techniques approved for clinical use in the UK are maternal spindle transfer (MST) and pronuclear transfer (PNT) (Box 2). Preclinical studies indicate that in both methods there may be some carry‐over of mutant mtDNA during the transfer of nDNA, but at a level unlikely to cause severe disease in the offspring.14 In the single live birth so far reported, using MST, the low levels of mtDNA found in newborn tissues were well below the threshold for risk of the particular mitochondrial disease.15
In Australia, the Prohibition of Human Cloning for Reproduction Act 2002 (the Act) prohibits the creation of a human embryo containing genetic material from more than two persons and bans the alteration of a genome of a human cell where that alteration is heritable through the germline. Since mtDNA is heritable through female gametes, the utilisation of a donor oocyte in mitochondrial donation means that genetic material from three persons is used to create the embryo (Box 2). However, mtDNA makes no contribution to the characteristics of an individual other than cellular bioenergetics. Moreover, unlike nDNA, mtDNA sequences are not unique to an individual but are shared by mothers and all their offspring, making mtDNA an identifier of families (through the maternal line) rather than individuals. There is a scientific view that a woman donating oocytes for mitochondrial donation does not contribute genetic material to the child’s unique genomic identity. Moreover, scientists consider that transfer of nDNA between oocytes does not constitute alteration of the genome, even though this replacement is heritable, since neither nDNA nor mtDNA are modified in any way. UK legislation refers specifically to nuclear DNA from the contributing eggs in the processes that are permitted under the regulation.10 The extensive consultation undertaken in Australia indicates mixed views on these matters. Maeve’s Law has been drafted to permit mitochondrial donation by exemption to the provisions of the Act, with strict regulation under a licensing framework to be administered by the NHMRC Embryo Research Licensing Committee.
The ethical and regulatory framework
While the potential benefits of mitochondrial donation for many families are well recognised, concerns remain about ethical risks. Apart from religious and moral views regarding interventions using assisted reproductive technologies in the formation of human life, the primary ethical concerns are about the rights of the children and oocyte donors and long term safety of the procedure for offspring born.13 Australia recognises the rights of children born from assisted reproductive technologies to know their genetic origins, and this right would extend to information about individuals donating oocytes for mitochondrial donation. Such access would need to be strictly controlled to protect the privacy of donors. While substantial pre‐clinical studies indicate the procedures are feasible and safe enough for clinical implementation, evidence of true efficacy is limited since only one live birth has so far been reported in the public domain. It is, however, recognised by scientists and clinicians that further technical refinements are needed to minimise the carry‐over of mutant mtDNA, as even very low levels of certain mtDNA mutations carry risks of severe or late onset disease in the individual or selective transmission to their offspring.4 As with any new medical technology, further testing and refinement can be achieved most effectively in a clinical setting. The UK Parliament took a cautionary approach in allowing the procedure in licensed centres for stringently selected high risk cases with a requirement for follow‐up and reporting of outcomes.11 A similar but even more cautious approach is being proposed for Australia. If passed by Parliament, Maeve’s Law will enable a staged implementation of mitochondrial donation, with licensing for research and training and a clinical trial over 10 years to provide evidence of safety and efficacy before approval is given for clinical use.
Australia has a long history in, and an excellent regulatory environment for, procedures involving assisted reproductive technologies, through both federal and state legislation. Clinics require accreditation through the Reproductive Technology Accreditation Committee and must comply with the NHMRC Ethical guidelines on the use of assisted reproductive technology in clinical practice and research.16 Applications to develop new procedures need approval by the NHMRC Embryo Research Licensing Committee, which would regulate licenses for mitochondrial donation, guided by clinical experts. Australia has the clinical expertise in mitochondrial disease to evaluate and select eligible families, and provide clinical oversight of mitochondrial donation. Eligible couples and oocyte donors will require expert counselling to ensure they do not have unrealistic expectations about the outcomes, understand the rights of any offspring to know their genetic history, and can make informed decisions regarding their reproductive options.
Conclusions
Mitochondrial donation can significantly reduce the risk of maternally inherited mitochondrial disease transmission to offspring and, for some families, provides their only option to have unaffected, genetically related children. Australia has the clinical and scientific expertise to introduce mitochondrial donation in a highly regulated environment, but requires changes in legislation to adopt this innovative technology, as proposed in the Maeve’s Law currently under parliamentary review. A cautionary, staged approach is being considered for implementation in Australia. Establishment of a coordinated network of clinics forming a national service would allow equitable access to the procedure and the clinical expertise necessary to evaluate patient outcomes, provide expert follow‐up, and support research and training in this important area.
Box 1 – Common clinical syndromes caused by maternally inherited mutations in mitochondrial DNA (mtDNA)*
Clinical syndrome |
Clinical phenotype |
Age of onset |
Common causative mtDNA mutations | ||||||||||||
Maternally inherited Leigh syndrome (MILS) |
Motor and intellectual developmental delay and neurological disability, early death (by 3 years) |
3–12 months |
MT ATP6 point mutation (m.8993T>G/C) in > 90% of mtDNA |
||||||||||||
Neurogenic weakness with ataxia and retinitis pigmentosa (NARP) |
Ataxia, pigmentary retinopathy, weakness, seizures, neuropathy |
Childhood or early adult life |
MT ATP6 point mutation (m.8993T>G/C) in 70–80% of mtDNA |
||||||||||||
Mitochondrial encephalopathy, lactic acidosis, stroke‐like episodes (MELAS) |
A broad spectrum of clinical phenotypes including stroke‐like episodes with encephalopathy, recurrent headaches, and seizures manifesting in severe cases. Variable presence of myopathy, deafness, endocrinopathy (eg, diabetes and short stature), ataxia, and early death (10–35 years) |
Originally described in childhood but can present across the lifespan |
MT TL1 point mutations (m.3243A>G in 80%, m3252A>G, m.3271T>C); MT TQ and NADH dehydrogenase subunit and ND5 point mutations (m.4332G>A, m.13513G>A) |
||||||||||||
Myoclonus, epilepsy, and ragged‐red fibres (MERRF) |
Stimulus‐sensitive myoclonus, generalised focal seizures, ataxia, cardiomyopathy, and/or lipomas. A minority of patients have progressive external ophthalmoplegia |
Adolescent or early adult life |
MT TK point mutations (m.8344A>G most common, m.8356T>C, m.8363G>A) and MT TH point mutation (m.12147G>A) |
||||||||||||
Chronic progressive external ophthalmoplegia (CPEO) |
Ptosis and ophthalmoparesis, frequent proximal myopathy and variable other clinical features such as ataxia and cardiac arrhythmias or cardiomyopathy |
Any age but more severe phenotype with younger onset |
Single deletions and MT TL1 and MT TK point mutations (including m.3243A>G, m.8344A>G) |
||||||||||||
Leber hereditary optic neuropathy (LHON) |
Subacute painless unilateral progressing to bilateral visual failure. May also have dystonia, cardiac pre‐excitation syndromes and, in rare cases, (usually females) demyelination (Harding disease) |
Typically in early adulthood, more common in males |
MT ND1, ND4 and ND6 point mutations (m.3460G>A, m.11778G>A, m.14484T>C) |
||||||||||||
* Some related clinical syndromes are caused by mutations in nuclear DNA (nDNA) but are not included in this table. | |||||||||||||||
Box 2 – Schematic drawing showing techniques of maternal spindle transfer (MST) and pronuclear transfer (PNT) to generate embryos unaffected by maternally inherited mitochondrial disease*
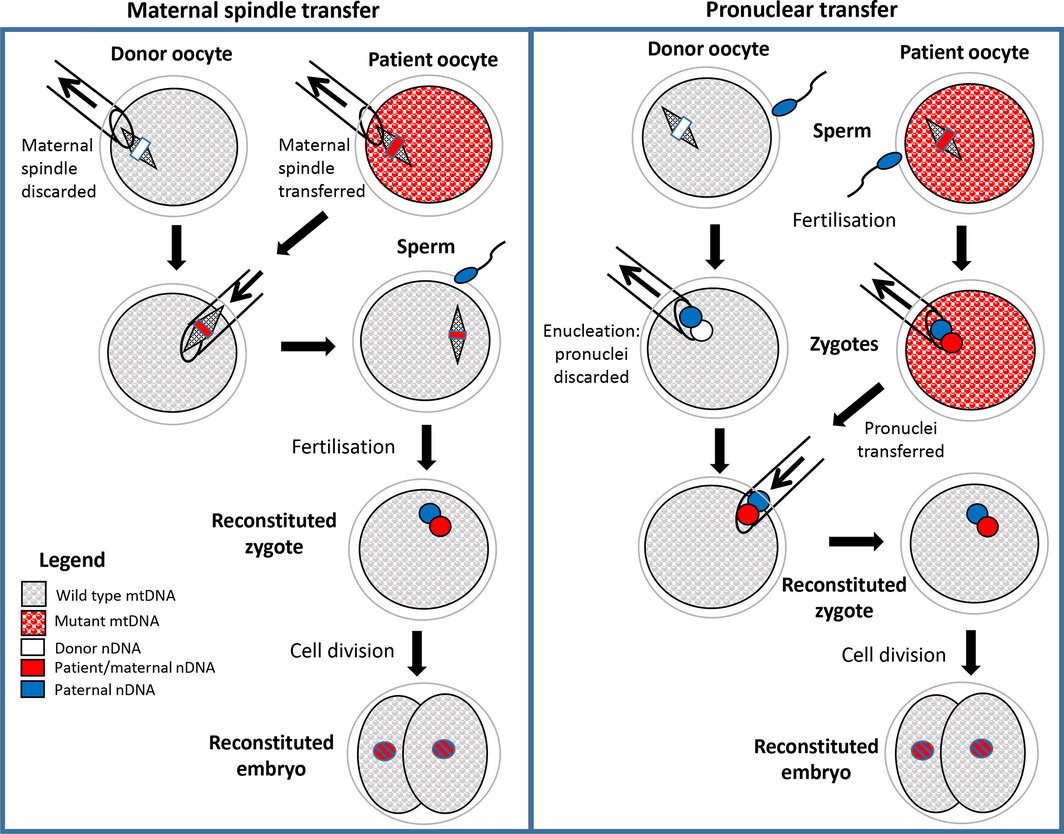
mtDNA = mitochondrial DNA; nDNA = nuclear DNA. * In both techniques, assisted reproductive technologies procedures are used to obtain mature oocytes from the female patient and the donor. In MST, the maternal spindles (including maternal chromosomes) are micropipetted out of each oocyte and the spindle from the patient oocyte is transferred to the donor oocyte, which is then fertilised to form a zygote. In PNT, both the patient and the donor oocytes are fertilised and the male and female pronuclei from the patient zygote are transferred to the enucleated donor zygote. In both MST and PNT, the resulting reconstituted zygote contains nDNA in the pronuclei from the patient’s oocyte and her partner’s (or donor) sperm and wild‐type mtDNA from the donor’s oocyte. The two pronuclei subsequently combine, and the division of the zygote into two cells marks the beginning of embryonic development. After several days in culture, embryos are transferred to the patient for subsequent development to term in utero.
Provenance: Not commissioned; externally peer reviewed.
- 1. Davis RL, Liang C, Sue CM. Mitochondrial diseases. Handb Clin Neurol 2018; 147: 125–141.
- 2. Wei W, Chinnery PF. Inheritance of mitochondrial DNA in humans: implications for rare and common diseases. J Int Med 2020; 287: 634–644.
- 3. Zaidi AA, Wilton PR, Shu‐Wei SuM, et al. Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc Natl Acad Sci USA 2019; 116: 25172–25178.
- 4. Sue CM. Mitochondrial disease: recognising more than just the tip of the iceberg. Med J Aust 2010; 193: 195–196. https://www.mja.com.au/journal/2010/193/4/mitochondrial‐disease‐recognising‐more‐just‐tip‐iceberg
- 5. Grady JP, Pickett SJ, Ng YS, et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol Med 2018; 10: e8262.
- 6. Otten ABC, Sallevelt SCEH, Carling PJ, et al. Mutation‐specific effects in germline transmission of pathogenic mtDNA variants. Hum Reprod 2018; 33: 1331–1341.
- 7. Rossignol R, Faustin B, Rocher C, et al. Mitochondrial threshold effects. Biochem J 2003; 370: 751–762.
- 8. Vandebona H, Mitchell P, Manwaring N, et al. Prevalence of mitochondrial 1555A→G mutation in adults of European descent. N Engl J Med 2009; 360: 642–644.
- 9. Pickett SJ, Blain A, Ng YS, et al. Mitochondrial donation — which women could benefit? N Engl J Med 2019; 380: 20.
- 10. Richardson J, Irving L, Hyslop LA, et al. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells 2015; 33: 639–645.
- 11. The Human Fertilisation and Embryology (Mitochondrial Donation) Regulations 2015. UK Statutory Instruments 2015 No. 572. https://www.legislation.gov.uk/uksi/2015/572/contents/made (viewed Aug 2021).
- 12. Newson AJ, de Lacey S, Dowling DK, et al. Public attitudes towards novel reproductive technologies: a citizens’ jury on mitochondrial donation. Hum Reprod 2019; 34: 751–757.
- 13. National Health and Medical Research Council. Report on NHMRC’s public consultation on the social and ethical issues raised by mitochondrial donation (consultation report). NHMRC 2020. https://www.nhmrc.gov.au/mitochondrial‐donation‐0#downlo (viewed Aug 2021).
- 14. Greenfield A, Braude P, Flinter F, et al. Assisted reproductive technologies to prevent human mitochondrial disease transmission. Nat Biotechnol 2017; 35: 1059–1068.
- 15. Zhang J, Liu H, Luo S, et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online 2017; 34: 361–368.
- 16. National Health and Medical Research Council. Ethical guidelines on the use of assisted reproductive technology in clinical practice and research. Canberra: NHMRC, 2017. https://www.nhmrc.gov.au/art (viewed Oct 2021).
Carolyn Sue is a Medical Research Future Fund National Health and Medical Research Council Practitioner Fellow (APP1136800).
No relevant disclosures.