





Volume 220 - Issue 2
Skin fragility disorder misdiagnosed as child abuse: a cautionary tale
Authors: Anneliese Willems, Lauren Weston and Susan Robertson
Med J Aust 2024; 220 (2): 71-73. || doi: 10.5694/mja2.52188
Published online: 5 February 2024
Published online: 5 February 2024
A two-year-old girl presented in distress after a minor fall in a sandpit at a childcare facility
Clinical record
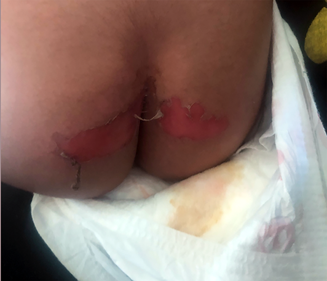
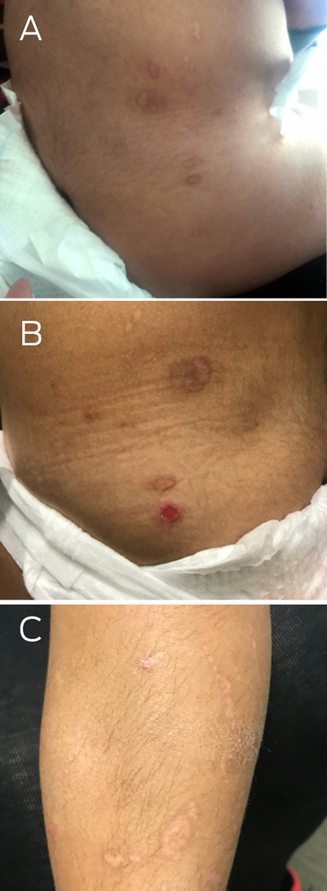
A two‐year‐old girl presented in distress after a minor fall in a sandpit at a childcare facility. On examination, she had large superficial erosions of the medial buttocks in a kissing distribution (Box 1). Further examination revealed multiple linear and round atrophic scars with post‐inflammatory hyperpigmentation affecting the trunk, left flank, buttocks and legs (Box 2, A–C). She was also noted to have an additional ten‐cent‐sized superficial erosion to her lower back (Box 2, B). There was no involvement of the head, neck and mucosae, and no deformity of the hair, teeth and nails. There was clinical concern that these erosions might represent non‐accidental injury (such as burns) and she was referred to a Forensic Paediatric Medical Service (FPMS) for further assessment.
The FPMS obtained collateral history from her parents and childcare providers that did not uncover any social concerns and revealed that she had suffered from intermittent spontaneous skin blisters and erosions mainly affecting the trunk and lower legs from two months of age. The FPMS sought dermatology consultation, who raised the suspicion for a skin fragility disorder. Further history revealed that there was no noted association with trauma and she had previously been diagnosed with recurrent skin infections. Once present, the blisters would self‐resolve within a few weeks, leaving residual scars and pigmentation. There was no family history of similar skin lesions. She was the first born to second cousins with known α‐thalassaemia trait but was otherwise well. The FPMS recommended investigation to exclude a skin fragility disorder and determined that there were no forensic concerns.
A 2mm skin biopsy was performed, under nitrous oxide and midazolam procedural sedation, to the left hip site where friction had been applied — produced by gently rubbing the skin for one minute, five minutes before taking the skin biopsy. The biopsy revealed an intraepidermal split with detached epidermis suggestive of epidermolysis bullosa simplex. Subsequently, genetic testing revealed homozygous pathogenic variants (c.5422C>T; p.(Arg1808*)) in the exophilin 5 gene (EXPH5), confirming EXPH5 autosomal recessive epidermolysis bullosa simplex. This is a rare subtype of epidermolysis bullosa with very few case reports in the literature and, to our knowledge, it has not previously been reported in the Australian population.1
This patient is currently managed through a multidisciplinary service for children with epidermolysis bullosa. She was enrolled in the federally funded National Epidermolysis Bullosa Dressings Scheme to support access to specialised non‐stick dressings and wound care supplies, which can be prohibitively costly to purchase privately,2 and her family linked in with a statewide epidermolysis bullosa health service and a patient support group for people living with epidermolysis bullosa. She has responded well to preventive treatment and has had fewer lesions appearing over time.
Discussion
Epidermolysis bullosa encompasses a heterogenous group of diseases that cause fragility and blistering to the skin and mucous membranes.3 Epidermolysis bullosa arises from inherited defects in the adhesion molecules that anchor the epidermis to the dermis. Epidermolysis bullosa is characterised by blisters and erosions, usually at the sites of friction or mild trauma such as the hands and feet.3 Normal everyday activities can result in blister formation, such as friction from a minor fall when wearing a nappy, as in this case.
There are four major types of epidermolysis bullosa: epidermolysis bullosa simplex, junctional epidermolysis bullosa, dystrophic epidermolysis bullosa and Kindler epidermolysis bullosa, distinguished by the layer of the skin within which the blisters form.3 Within these types there are multiple clinical subtypes exhibiting a vast spectrum of severity. Severe subtypes (particularly recessive dystrophic epidermolysis bullosa and junctional epidermolysis bullosa) usually present at birth and can be life‐limiting with the development of multiple extracutaneous complications including cutaneous squamous cell carcinoma.3 Milder subtypes may present later and have a normal life expectancy with fewer complications, such as our patient. Epidermolysis bullosa has traditionally been diagnosed by skin biopsy of a newly induced blister, which undergoes histology, immunofluorescence antigen mapping and transmission electron microscopy to elucidate the level of skin cleavage and identify deficiency of hemidesmosome proteins.3 Increasingly, with better access to genetic testing, genotyping has become the preferred method of diagnosis, particularly as the biopsy technique is less reliable for diagnosis of milder epidermolysis bullosa subtypes.4
As the history of skin fragility was subtle, our case presented in a manner that could be interpreted as child abuse. Epidermolysis bullosa is rare, the Australian prevalence is 10.3 cases per million population.5 Non‐accidental injuries in children are unfortunately vastly more prevalent. In 2021–22, about 178000 children aged 0–17 years received child protection services in Australia, and eight per 1000 children were considered to have substantiated physical abuse or neglect.6 With such prevalence, doctors have an ethical and legal mandate to remain vigilant for signs of possible non‐accidental injury in children. Yet many genuine skin diseases, as in this case, may present with cutaneous stigmata that could be interpreted as non‐accidental injury. Potential mimics for child abuse are broad and include genuine accidental trauma, coagulation disorders, connective tissue diseases, inflammatory disorders, artifactual skin findings and, as in this case, disorders of skin fragility.7 It is important to keep an open mind as misdiagnosis of child abuse can result in significant distress for families as well as delay treatment for the underlying condition. Clinician awareness of and vigilance for these possible diagnoses can aid in having a less traumatic diagnostic journey for families.
This case highlights a story of an unexplained injury with a distressed child that was ultimately referred for forensic assessment. Through keeping an open mind, including openness in the clinical discourse and careful collateral history and cross‐specialty care, a diagnosis of a skin fragility disorder was able to be made revealing this was not, in fact, a case of child abuse.
There is currently no cure for epidermolysis bullosa. Management is largely preventive skin care to reduce blistering, including avoidance of friction to the skin, liberal emollient application, prompt puncturing of blisters and the use of antibacterial agents and non‐adherent dressings for active lesions to prevent infection, reduce pain and assist wound healing.8 Children with suspected skin fragility disorders should be referred for specialist assessment to secure an accurate diagnosis and enable families to access necessary support.
Lessons from practice
- Unexplained skin injuries in children may have underlying causes beyond physical abuse, such as rare skin fragility disorders like epidermolysis bullosa.
- Child abuse mimics are diverse and can include genuine accidental trauma, connective tissue disorders, and inflammatory conditions, highlighting the importance of keeping an open mind during assessment.
- Thorough collateral history from parents or caregivers is essential in assessing unusual skin presentations in children.
- Children with suspected skin fragility disorders should be referred for specialist assessment to secure an accurate diagnosis and, if eligible, facilitate access to necessary supports, including the government‐funded National Epidermolysis Bullosa Dressing Scheme and patient support groups.
Competing interests
No relevant disclosures.
References
- Pigors M, Schwieger‐Briel A, Leppert J, et al. Molecular heterogeneity of epidermolysis bullosa simplex: contribution of EXPH5 mutations. J Invest Dermatol 2014; 134: 842‐845.
- Independence Australia Group. Welcome to the National Epidermolysis Bullosa (EB) Dressing Scheme 2021. https://www.ebdressings.com.au/ (viewed Aug 2023).
- Bardhan A, Bruckner‐Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers 2020; 6: 78.
- Saunderson RB, Vekic DA, Mallitt K, et al. A retrospective cohort study evaluating the accuracy of clinical diagnosis compared with immunofluorescence and electron microscopy in children with inherited epidermolysis bullosa. Br J Dermatol 2019; 180: 1258‐1259.
- Kho YC, Rhodes LM, Robertson SJ, et al. Epidemiology of epidermolysis bullosa in the antipodes: the Australasian Epidermolysis Bullosa Registry with a focus on Herlitz junctional epidermolysis bullosa. Arch Dermatol 2010; 146: 635‐640.
- Australian Institute of Health and Welfare. Child protection Australia 2021–22 [website]. Canberra: AIHW, 2023. https://www.aihw.gov.au/reports/child‐protection/child‐protection‐australia‐2021‐22/contents/child‐protection‐system‐in‐australia (viewed July 2023).
- Schwartz KA, Metz J, Feldman K, Sidbury R, Lindberg DM. Cutaneous findings mistaken for physical abuse: present but not pervasive. Pediatr Dermatol 2014; https://doi.org/10.1111/pde.12290 [Epub ahead of print].
- Denyer J, Pillay E, Clapham J. Best practice guidelines for skin and wound care in epidermolysis bullosa. An international consensus. London: Wounds International, 2017. https://woundsinternational.com/wp‐content/uploads/sites/8/2023/02/79912622fffa0956d1619feb123f35ed.pdf (viewed Nov 2023).
Provenance: Not commissioned; externally peer reviewed.