





Perspectives
Open access CC BY NC ND
Volume 217 - Issue 2
Clinical gene technology in Australia: building on solid foundations
Authors: Gabrielle O'Sullivan, Joshua G Philips and John EJ Rasko
Med J Aust 2022; 217 (2): 65-70. || doi: 10.5694/mja2.51629
Published online: 18 July 2022
Published online: 18 July 2022
Many technical and regulatory hurdles have been overcome, but the field has a long way to go
Many technical and regulatory hurdles have been overcome, but the field has a long way to go
Who would have thought a small piece of messenger RNA (mRNA) wrapped in a delectable lipid could do so much? Coronavirus disease 2019 (COVID‐19) has transfixed public attention and reminded us that gene technology is essential for vaccine development. Indeed, successful clinical implementation requires convergent input from research, manufacturing, clinical trials, regulation, supply chains, commerce, health and social systems, and clear definitions to counter misinformation (Box 1).
Although clinical gene therapy designed to cure human disorders remains relatively new, there have been a series of advances since the first successful clinical trial in 1990,1 and there are strong indicators to suggest an enormous acceleration in its growth.2 For example, there are over 900 investigational new drugs for ongoing clinical studies, and the United States Food and Drug Administration (FDA) anticipates ten to 20 new product approvals per year by 2025.3 Global investment in regenerative medicine, which includes cell and gene therapies and tissue‐engineered products, increased by almost 50% from US$13.5 billion in 2018 to US$19.9 billion in 2020, with 1200 ongoing clinical trials.4 It is anticipated that the global market will reach AU$120 billion by 2035 and that Australia could potentially achieve AU$6 billion in annual revenue, predominantly in exports, and gain 6000 jobs and earlier access to new treatments.5 Currently, there are 153 clinical trials6 and nine approved gene therapies in Australia, such as Spinraza (Biogen), Kymriah (Novartis), Yescarta (Kite Pharma), Zolgensma (Novartis) and Luxturna (Novartis) (Box 2).
Notwithstanding the successes, these therapies are complex and there are many open questions about long term safety, efficacy and durability that need more time to answer. At the core of these new therapies is the fact that they are designed to modify living cells, often persisting for the lifetime of the recipient. The benefits, risks and complexities arise from the biological nature of the treatments; for example, but not limited to, gene modified cells, viruses, viral vectors, DNA and RNA. Furthermore, how they are introduced into the body, directly (via in vivo gene therapy) or indirectly (via ex vivo gene therapy, whereby cells are engineered in the laboratory and then infused into the patient); how they enter cells; where they act; and how they interact with the human genome (Box 3) all have an impact. As a result, unlike most chemically synthesised drugs, these products are sensitive to degradation and contamination, and their clinically active components are not always clearly identified. Therefore, they are often defined by the processes and equipment used to make them. Changes to those processes may require additional clinical studies to demonstrate that their safety, identity and purity have not been compromised.
It is just over 20years since Australia’s national gene technology regulatory scheme administered by the Office of the Gene Technology Regulator (OGTR) came into effect.7 The OGTR and the Therapeutic Goods Administration (TGA) are integral to regulatory approvals for gene therapies in Australia. It is an opportune time to provide a snapshot of the evidence for using gene therapy in a clinical setting and what the setbacks to delivery are. Gene therapy for haemophilia, chimeric antigen receptor T (CAR‐T) cell therapy for cancer, and gene editing illustrate many of the issues.
Haemophilia
The inherited bleeding disorder haemophilia presents a compelling candidate for gene therapy because it is caused by mutation of a single gene — the gene for clotting factor VIII in haemophilia A, or clotting factor IX in haemophilia B. A modest increase in the level of circulating clotting factor (≥5% of normal) in severe haemophilia, can prevent much of the disease morbidity and mortality.8 Haemophilia has an enormous impact on patients’ lives. Depending on the severity, it can cause painful recurrent spontaneous bleeding into joints and require lifelong injections of clotting factor concentrate.
Gene therapy for haemophilia is entering the late clinical trial stage, with at least ten ongoing trials for haemophilia A and six for haemophilia B.9 This form of in vivo gene therapy involves a single intravenous injection of an adeno‐associated virus (AAV) vector that has been engineered to home to the liver and produce the required clotting factor there for the remainder of an adult’s life.
Sustained therapeutic levels of clotting factor activity above 20% and 5–36% of normal have been demonstrated for haemophilia A and B respectively.8,10 Clinically impressive reductions in annual bleeding rate (by 83.8% for haemophilia A,11 and by 98% for haemophilia B8) and factor replacement usage (by 98.6% for haemophilia A11 and by 66%12 and 80%13 for haemophilia B) have been observed.
However, durability of the therapeutic effect has not yet been adequately demonstrated to achieve marketing approval. In mid‐2020, many expected BioMarin’s gene therapy for severe haemophilia A (valoctocogene roxaparvovec) to become the first US FDA‐approved haemophilia gene therapy. However, 3‐year results from the phase 1/2 trial showed a decline in factor VIII levels 12 months after infusion.10 This led the FDA to conclude that 2‐year instead of 6‐month phase 3 data supporting durability of effect would be required for approval, despite favourable clinical results in all study phases.14
It remains unclear whether AAV vector gene therapy for haemophilia will be a once‐off treatment for life. Because AAV vector DNA predominantly exists as independent episomes in the cell nucleus, gradual loss during normal liver cell turnover, and patient immune responses to the vector coating (capsid), may limit the durability of clinical benefit. Natural pre‐existing immunity to wild‐type AAV vectors may also reduce access to AAV vector‐based therapies if not properly identified.
Despite a history of safe and beneficial use in many human clinical trials, it is also unclear how safe AAV vector gene therapy will be in the long term and what factors affect patient safety.15,16 In September 2021, dosing was suspended in two AAV vector clinical trials pending investigations into implications for human therapy: one by Astellas for X‐linked myotubular myopathy which witnessed four deaths over 2020–2021, and the other by BioMarin for phenylketonuria due to reports of liver tumours in mice in preclinical studies.17 Also in September 2021, the FDA held a 2‐day meeting that examined AAV vector‐gene therapy cancer risk and liver and neurological toxicities. The expert panel concluded that improvements to research tools, patient screening, side‐effect monitoring, production standards, risk assessments and scrutiny of components are needed. They also indicated that longer term animal experiments and follow‐up of over 800 patients treated with Zolgensma — an approved AAV vector‐gene therapy for spinal muscular atrophy — would provide useful information. However, it did not recommend major changes to the way studies are conducted, upper dose limits, or slowing down the pace of research.18 Real world evidence is also important for regulatory decision making and for monitoring post‐market safety.
CAR‐T cell therapy for blood cancers
CAR‐T cell therapies have achieved exceptional clinical outcomes in specific cancer patient groups (an area of large unmet need). In particular, CAR‐T cell immunogene therapies that target CD19 on B cells have achieved complete response and remission rates of 77% and 80% in acute lymphoblastic leukaemia.19,20
Traditional CAR‐T cell therapies are manufactured by collecting peripheral blood T cells from patients with B‐cell malignancies and genetically engineering them in a specialised laboratory to express a receptor (such as the CD19 receptor). When reinfused back into the patient, the engineered cells target an antigen on the malignant cells (such as CD19) and initiate an antitumour immune response.
Despite their success, implementing CAR‐T cell therapy is complex and there are many technical and organisational hurdles to overcome. About 26% of treated patients develop cytokine release syndrome and 12% develop neurotoxicity, both grade 3 or above,20 which can be life‐threatening and require intensive care. In addition, the cells can become exhausted and ineffective due to overstimulation by the target antigens. Clinicians have refined toxicity management options, and novel CAR‐T cell designs, combined with improved potency assays, may help reduce complications in the future.
Other hurdles are the high cost of the therapies (currently over AU$500000 per patient without government funding) and the extensive multidisciplinary specialist infrastructure needed for safe delivery to patients.21 Currently, two CAR‐T cell therapies for B‐cell cancers (Kymriah and Yescarta) are funded for delivery in Australian public hospitals through a 50/50 federal and state government funding arrangement in registered accredited hospitals.22 Prospective payments are made to the service providers based on annual submissions of actual costs to the Independent Hospital Pricing Authority (IHPA). This has been vital in enabling Australians to gain early access to these therapies and data collection for improvements. However, because the services are complex and data collection only commenced in 2020, a full understanding of the costs and scope has yet to develop. Some gaps have been identified. For example, IHPA noted advice from jurisdictions “that the Relative Value Units for pathology, and used for allocating costs associated with bed days do not adequately account for the intensive nature of CAR‐T treatment”.22 It will take time, and increased patient numbers and data, to be able to move from retrospective reconciled funding to more concurrent activity‐based funding.
Gene editing
Gene editing using targetable enzymes (nucleases) that cut DNA at precise locations and allow the deletion, insertion or correction of genes have been hailed as revolutionary and are being used in successful clinical trials. For example, the Nobel Prize‐winning technology CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 is applied to create “off‐the‐shelf” CAR‐T cells in clinical trials for B‐cell malignancies and solid tumours. In this case, T cells from a healthy universal donor are used instead of T cells from the patient. Recently, the first in vivo CRISPR/Cas9 gene editing human clinical study was conducted for patients with transthyretin amyloidosis. Significant reductions in serum transthyretin protein, which accumulates in a misfolded form in patients’ tissues, were reported.23 The US FDA has also approved a clinical trial for another in vivo CRISPR study in human immunodeficiency virus (HIV)‐infected individuals.24 However, gene editing also presents challenges that remain to be solved, such as potential off‐target effects and damage to DNA. Recently, a clinical trial of a gene edited “off‐the‐shelf” CAR‐T cell therapy for lymphoma was halted until an investigation concluded that a chromosomal abnormality detected in a patient after treatment was unrelated to gene editing or manufacturing processes.25 It is widely accepted that better research tools are need for detecting off‐target effects.
Other issues to be resolved
Despite their clinically transformative effects, approaches to the valuation and reimbursement of gene therapies are still being worked out globally, and it can be difficult for companies to reconcile payment for their products. Bluebird bio withdrew two approved therapies — Zynteglo for β‐thalassaemia and Skysona for cerebral adrenoleukodystrophy — from the European market after failing to obtain payer agreements.26 Depending on the therapy, the target disorder, and population, a range of innovative approaches will be needed to resolve the challenges of return on investment versus equity in access. Such approaches may include outcomes‐, indication‐ and milestone‐based reimbursement in addition to improved discounting and backup funding for health technology assessments.27,28
In Australia there is a serious shortage of advanced therapy manufacturing and Good Manufacturing Practice (GMP) workforce skills,29 and lack of a coordinated approach to collecting and reporting patient data.30 These limit Australia’s ability to manufacture advanced therapeutics and plan research, treatments and clinical services.30 Increasing efforts by state and federal governments as well as institutions and commercial for‐profit entities are required to develop a robust approach to local advanced therapy manufacturing capacities.
Global regulatory harmonisation of gene therapy definitions, terminology, GMP standards and timeframes for approval of clinical trials and marketing approvals will be further required to overcome limitations.
Reviews of the gene technology sector have been undertaken to improve access in Australia. These include the review into the national gene technology regulatory scheme,31 the Inquiry into approval processes for new drugs and novel medical technologies in Australia32 and the Review of the National Medicines Policy.33
Conclusion
The implementation of gene technology in the clinical setting has already provided enormous benefits to human health. A rapid increase in approvals for gene therapies is anticipated. However, the field is still new, the therapies are biologically active and very complex, and the mechanisms for safe production and delivery to patients are also complex and still developing. Careful planning, ongoing research and longer term data are needed to overcome current limitations across many areas.
Box 1 – Definitions and key concepts
|
|
Definitions |
||||||||||||||
|
|
|||||||||||||||
|
Gene technology |
Technology that is applied to directly modify existing genetic material, such as cellular DNA, in a specific way or to make new genetic material. It is also known as genetic engineering or genetic modification and can be used to add, alter or remove genes from cellular genomes. The technology can be used in the manufacture and application of vaccines and gene therapies. |
||||||||||||||
|
Gene therapy |
A technique for modifying a patient’s genes to treat a disease. This could be by replacing or inactivating a faulty gene or by adding a therapeutic gene. Viral vectors may be used to deliver therapeutic genes into patients directly, such as in adeno‐associated viral vector‐based gene therapy for haemophilia, or indirectly, whereby cells are collected from a donor and modified ex vivo before being administered to a recipient, such as gene modified cell therapy (eg, CAR‐T cell therapy, see below). |
||||||||||||||
|
Gene modified cell therapy |
The treatment of a patient with cells that have been collected from a donor and modified ex vivo (eg, using gene therapy or gene editing techniques) for administration to the patient (eg, CAR‐T cell therapy for cancer). |
||||||||||||||
|
Gene editing |
Techniques for making precise edits to the genome using enzymes. The most widely known is the CRISPR/Cas9 system in which the enzyme (Cas9) is complexed with a synthetic guide RNA that directs the enzyme to a target site in the genome where it cleaves specific DNA sequences and allows sequences to be added, removed or altered in situ (ie, “edited”). |
||||||||||||||
|
mRNA technology and vaccines |
DNA is the cellular store of genetic information that allows proteins to be made by cells. In order for the information in the DNA to be used to make a specific protein, the information for that protein is transcribed into a transient intermediary molecule known as mRNA. In cells, mRNA is transported out of the nucleus and into the cytoplasm where it is used as a template by the cell’s own machinery to translate the mRNA to make the protein. In a clinical setting, mRNA can be delivered into cells as transient templates which cells can use to make proteins (eg, the Comirnaty [Pfizer] mRNA vaccine for COVID‐19, which the cell uses as a template to make immunising spike protein). mRNA vaccines do not enter the cell nucleus and do not modify cellular DNA and are not gene therapy. |
||||||||||||||
|
|
|||||||||||||||
|
CAR = chimeric antigen receptor; COVID‐19 = coronavirus disease 2019; mRNA = messenger RNA. |
|||||||||||||||
Box 2 – Snapshot of gene therapy since 1990
|
Gene therapy snapshot |
TGA‐approved therapies* |
||||||||||||||
|
Globally |
Australia |
Product |
Type of therapy |
Indication |
Funding in Australia |
OGTR‐licenced† |
|||||||||
|
|
|||||||||||||||
|
Imlygic (Amgen) |
Oncolytic gene‐modified viral gene therapy |
Melanoma |
Assessed by PBAC but not recommended for public funding on PBS (July 2016) |
Yes |
|||||||||||
|
Spinraza (Biogen) |
RNA therapy |
Spinal muscular atrophy |
Publicly funded on PBS |
Not required |
|||||||||||
|
Kymriah (Novartis) |
CAR‐T cell therapy |
Specific B‐cell cancers |
Publicly funded jointly by the Australian Government and states via NHRA |
Not required |
|||||||||||
|
Yescarta (Kite Pharma) |
CAR‐T cell therapy |
Specific B‐cell cancers |
Publicly funded via NHRA |
Not required |
|||||||||||
|
Zolgensma (Novartis) |
AAV vector gene therapy |
Spinal muscular atrophy |
Publicly funded on PBS |
Yes |
|||||||||||
|
Luxturna (Novartis) |
AAV vector gene therapy |
Inherited retinal dystrophy |
Publicly funded via NHRA |
Yes |
|||||||||||
|
Evrysdi (Roche Genentech) |
RNA therapy |
Spinal muscular atrophy |
Publicly funded on PBS |
Not required |
|||||||||||
|
Tecartus (Kite Pharma) |
CAR‐T cell therapy |
Mantle cell lymphoma |
Recommended by MSAC for public funding via NHRA (July 2021) |
Not required |
|||||||||||
|
Leqvio (Novartis) |
RNA therapy |
Cardiovascular disease |
Not currently funding |
Not required |
|||||||||||
|
|
|||||||||||||||
|
AAV = adeno‐associated virus; CAR = chimeric antigen receptor; MSAC = Medical Services Advisory Committee; NHRA = National Health Reform Arrangements; OGTR = Office of the Gene Technology Regulator; PBAC = Pharmaceutical Benefits Advisory Committee; PBS = Pharmaceutical Benefits Scheme; TGA = Therapeutic Goods Administration. * The therapies listed here are TGA‐approved and on the Australian Register of Therapeutic Goods (ARTG). Therapeutic goods listed on the ARTG can be lawfully supplied in Australia for the treatment of specified disorders in accordance with specified conditions of approval. The broader health technology of CAR‐T cell therapy is not eligible for funding through the Medical Benefits Schedule or the PBS. Instead, CAR‐T cell therapies are assessed by the MSAC, which advises the Australian Government on whether a new medical service should be publicly funded. If recommended by MSAC, the therapy is jointly funded by the Australian Government and the states for specific indications under the NHRA. ARTG‐listed gene therapies and RNA therapies classed as prescription medicines are assessed by the PBAC and, if recommended as suitable, are funded through a listing on the PBS. Luxturna is an exception to this, as it was assessed by MSAC. † OGTR‐licenced means the supply and use of the therapy are covered by a licence from the OGTR. In Australia, the use of viruses or viral vectors modified by gene technology require an OGTR licence. OGTR licences specify risk management requirements for managing potential environmental and health risks from gene‐modified viruses and viral vectors. The use of CAR‐T cell and RNA therapy does not have such environmental risks and therefore does not require an OGTR licence. |
|||||||||||||||
Box 3 – Features of different genetic material delivery methods
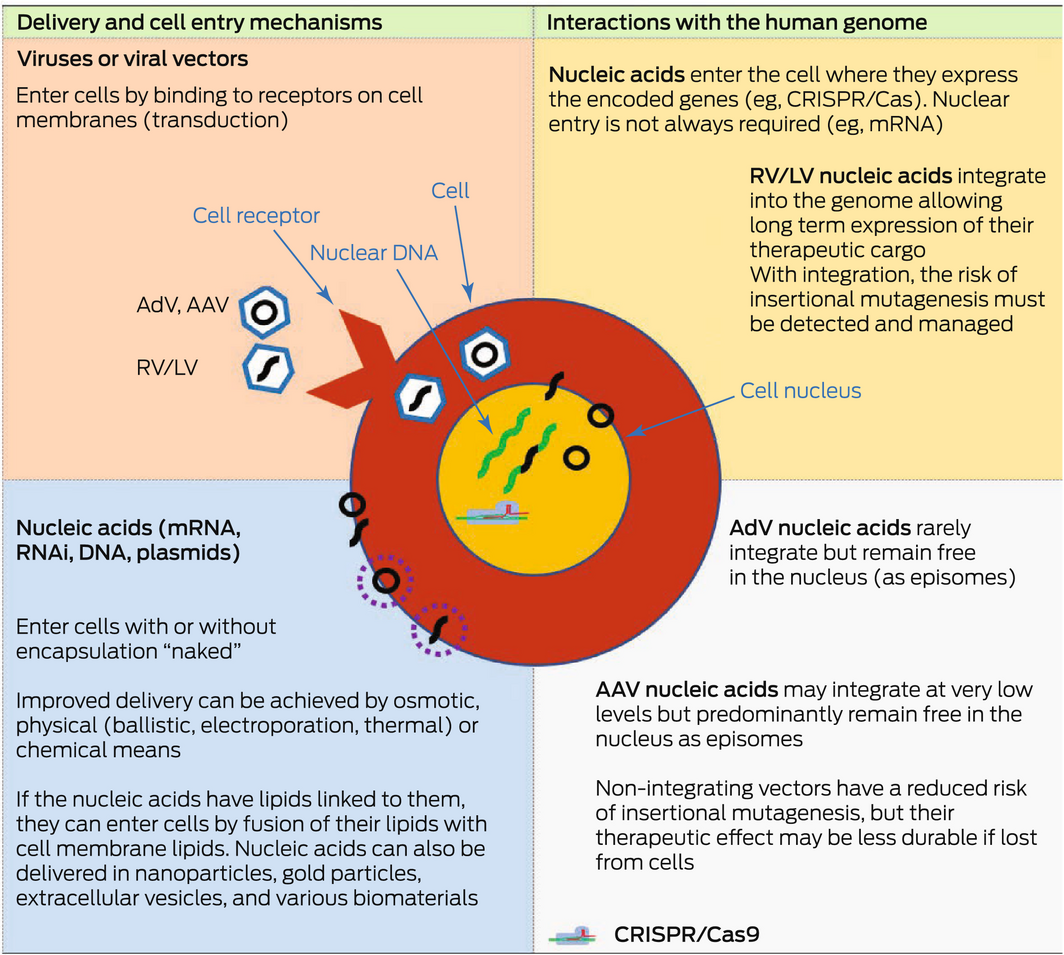
AAV = adeno‐associated virus; AdV = adenoviral vector; Cas9 = CRISPR‐associated protein 9; CRISPR = Clustered Regularly Interspaced Short Palindromic Repeats; LV = lentiviral vector; mRNA = messenger RNA; RNAi = RNA interference; RV = retroviral vector. Green strands represent the endogenous nuclear DNA in the cell. Black strands represent exogenous nucleic acids delivered into the cell via the various delivery mechanisms shown. They can be circular or non‐circular molecules. Stippled circles represent various types of coatings that can be used to encapsulate exogenous nucleic acids to assist their delivery into cells. Nucleic acids may be delivered directly into the cell or via the use of viral vectors. Viral vectors such as AAV vectors have been widely used as gene therapy vectors; for example, in haemophilia clinical trials and in the treatment of inherited blindness (Luxturna; Novartis). AdVs and mRNAs have been used as vaccines, such as the coronavirus disease 2019 (COVID‐19) vaccines. Lentiviral vectors are a subclass of retroviral vectors and have been used ex vivo to modify cells for the treatment of β‐thalassaemia and sickle cell disease. CRISPR/Cas9 has also been used ex vivo to create chimeric antigen receptor T (CAR‐T) cells that target particular cancers (eg, B‐cell malignancies).
Competing interests
John Rasko has served as Chair on the Therapeutic Goods Administration Advisory Committee on Biologicals and the National Health and Medical Research Council Mitochondrial Donation Expert Working Committee. As Head of Department, Cell and Molecular Therapies, Royal Prince Alfred Hospital (RPAH), and as Head of the Gene and Stem Cell Program at the Centenary Institute, he is responsible for undertaking investigator and pharmaceutical companies‐sponsored clinical trials of immunotherapies, cellular therapies and gene therapies at RPAH and basic research at the Centenary Institute, often in collaboration with biotechnology and infrastructure companies. He has also had Material Transfer Agreements, consultancy or honoraria with a number of companies, including Gilead, Novartis, Bluebird bio, Spark Therapeutics, Cynata, Pfizer, Atara Bio, Bayer and CRISPR Therapeutics. Any patents are held by the Sydney Local Health District, the Centenary Institute or the University of Sydney as his primary affiliations. He has extensive research and clinical interests in gene technology, especially in haemophilia gene therapy and CAR‐T cells. Gabrielle O’Sullivan has served on the Gene Technology Technical Advisory Committee since 2011, including as cross‐member of the Gene Technology Ethics and Community Consultative Committee from 2015 to the present. Joshua Philips is employed by the Office of the Gene Technology Regulator.
Acknowledgements
John Rasko acknowledges grant funding from the National Health and Medical Research Council (Investigator Grant 1177305) “Driving clinical cell and gene therapy in Australia” and from Therapeutic Innovation Australia. These funding sources support John Rasko in developing the fields of cell and gene therapy in Australia, and had no specific role in this submitted work. We thank Michael Dornbusch and Heidi Mitchell of the Office of the Gene Technology Regulator for providing advice and guiding the development of this article. We also thank Charles Bailey and Bijay Dhungel of the Gene and Stem Cell Therapy Program at Centenary Institute for providing advice and assistance with the illustrations. The views expressed in the article are those of the authors alone and do not represent an official view of the Australian Government, the Gene Technology Regulator, New South Wales Health, the Sydney Local Health District or the Royal Prince Alfred Hospital.
References
- Anderson WF. Human gene therapy. Science 1992; 256: 808‐813.
- American Society of Gene and Cell Therapy. Gene, cell, and RNA therapy landscape: Q1 2022 quarterly data report: ASCGT, 2022. https://asgct.org/global/documents/asgct‐pharma‐intelligence‐q1‐2022‐report.aspx (viewed June 2022).
- US Food and Drug Administration. FDA continues strong support of innovation in development of gene therapy products: guidances issued today provide regulatory clarity for product developers [press release]. 28 Jan 2020. https://www.fda.gov/news‐events/press‐announcements/fda‐continues‐strong‐support‐innovation‐development‐gene‐therapy‐products (viewed June 2022).
- Alliance for Regenerative Medicine. 2020: Growth and resilience in regenerative medicine — annual report. ARM, 2020. http://alliancerm.org/wp‐content/uploads/2021/03/ARM_AR2020_FINAL‐PDF.pdf (viewed June 2022).
- MTP Connect. Regenerative medicine: opportunities for Australia — October 2018. MTP Connect, 2018. https://www.mtpconnect.org.au/Attachment?Action=Download&Attachment_id=104 (viewed June 2022).
- AusBiotech. Australia’s regenerative medicine clinical trials database. AusBiotech, 2021. https://www.ausbiotech.org/documents/item/667 (viewed June 2022).
- O'Sullivan GM, Philips JG, Mitchell HJ, et al. 20Years of legislation — how Australia has responded to the challenge of regulating genetically modified organisms in the clinic. Front Med (Lausanne) 2022; 9: 883434.
- Nathwani AC. Gene therapy for hemophilia. Hematology Am Soc Hematol Educ Program 2019; 1: 1‐8.
- Batty P, Lillicrap D. Hemophilia gene therapy: approaching the first licensed product. Hemasphere 2021; 5: e540.
- Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow‐up of AAV5‐hFVIII‐SQ gene therapy for hemophilia A. N Engl J Med 2020; 382: 29‐40.
- Ozelo MC, Mahlangu J, Pasi J, et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N Engl J Med 2022; 386: 1013‐1025.
- Nathwani AC, Tuddenham EGD, Rangarajan S, et al. Adenovirus‐associated virus vector‐mediated gene transfer in hemophilia B. N Engl J Med 2011; 365: 2357‐2365.
- George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high‐specific‐activity factor IX variant. N Engl J Med 2017; 377: 2215‐2227.
- Pierce GF. Gene therapy for hemophilia: are expectations matching reality? Mol Ther 2020; 28: 2097‐2098.
- George LA, Ragni MV, Rasko JEJ. Long‐term follow‐up of the first in human intravascular delivery of AAV for gene transfer: AAV2‐hFIX16 for severe hemophilia B. Mol Ther 2020; 28: 2073‐2082.
- Gene therapy at the crossroads [editorial]. Nat Biotechnol 2022; 40: 621.
- Philippidis A. Fourth boy dies in clinical trial of Astellas’ AT132. Hum Gene Ther 2021; 32: 1008‐1010.
- Pagliarulo N, Fidler B. At FDA meeting, gene therapy experts wrestle with field’s blindspots. BiopharmaDive 2021; 3 Sept. https://www.biopharmadive.com/news/fda‐gene‐therapy‐meeting‐safety‐cancer‐liver/606088/ (viewed June 2022).
- Grigor EJM, Fergusson D, Kekre N, et al. Risks and benefits of chimeric antigen receptor T‐cell (CAR‐T) therapy in cancer: a systematic review and meta‐analysis. Transfus Med Rev 2019; 33: 98‐110.
- Anagnostou T, Riaz IB, Hashmi SK, et al. Anti‐CD19 chimeric antigen receptor T‐cell therapy in acute lymphocytic leukaemia: a systematic review and meta‐analysis. Lancet Haematol 2020; 7: e816‐e826.
- Velickovic ZM, Rasko JEJ. Establishing a robust chimeric antigen receptor T‐cell therapy program in Australia: the Royal Prince Alfred Hospital experience. Cytotherapy 2022; 24: 45‐48.
- Independent Hospital Pricing Authority. Chimeric antigen receptor T cell therapy (CAR‐T cell): guidelines for costing, counting and reconciliation of funding. IHPA, 2020. https://www.ihpa.gov.au/sites/default/files/publications/chimeric_antigen_receptor_t_cell_therapy_car‐t_cell_guidelines_for_costing_counting_and_reconciliation_of_funding.pdf (viewed June 2022).
- Gillmore JD, Gane E, Taubel J, et al. CRISPR‐Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med 2021; 385: 493‐502.
- Excision BioTherapeutics. Excision receives FDA clearance of IND for phase 1/2 trial of EBT‐101 CRISPR‐based therapeutic for treatment of HIV. GlobeNewswire 2021; 15 Sept. https://www.globenewswire.com/news‐release/2021/09/15/2297456/0/en/Excision‐Receives‐FDA‐Clearance‐of‐IND‐for‐Phase‐1‐2‐Trial‐of‐EBT‐101‐CRISPR‐Based‐Therapeutic‐for‐Treatment‐of‐HIV.html (viewed June 2022).
- Allogene Therapeutics. Allogene Therapeutics announces removal of FDA clinical hold across all AlloCAR T clinical trials [press release]. 10 Jan 2022. https://ir.allogene.com/news‐releases/news‐release‐details/allogene‐therapeutics‐announces‐removal‐fda‐clinical‐hold‐across (viewed June 2022).
- Pagliarulo N. Bluebird, winding down in Europe, withdraws another rare disease gene therapy. BiopharmaDive 2021; 21 Oct. https://www.biopharmadive.com/news/bluebird‐withdraw‐gene‐therapy‐europe‐skysona/608666/ (viewed June 2022).
- Underwood G. Finding the right reimbursement model for gene therapies. Pharmaphorum 2019; 23 July. https://pharmaphorum.com/views‐and‐analysis/finding‐the‐right‐reimbursement‐model‐for‐gene‐therapies/ (viewed June 2022).
- Jørgensena J, Hannab E, Kefalas P. Outcomes‐based reimbursement for gene therapies in practice: the experience of recently launched CAR‐T cell therapies in major European countries. J Mark Access Health Policy 2020; 8: 1715536.
- AusBiotech. Australia’s regenerative medicine manufacturing capability and capacity. AusBiotech, 2021. https://www.ausbiotech.org/documents/item/666 (viewed June 2022).
- AusBiotech. The regenerative medicine value chain: the pathway from discovery to patient delivery. AusBiotech, 2021. https://www.ausbiotech.org/documents/item/664 (viewed June 2022).
- Australian Government, Department of Health. Explanatory paper on the Consultation Regulation Impact Statement (CRIS), modernising and future‐proofing the National Gene Technology Scheme: proposed regulatory framework to support implementation of the Third Review of the Scheme, December 2020. https://www.genetechnology.gov.au/reviews‐and‐consultations/past/2017‐third‐review?utm_source=health.gov.au&utm_medium=callout‐auto‐custom&utm_campaign=digital_transformation (viewed June 2022).
- House of Representatives Standing Committee on Health, Aged Care and Sport. The new frontier — delivering better health for all Australians: inquiry into approval processes for new drugs and novel medical technologies in Australia. Canberra: Commonwealth of Australia, 2021. https://www.aph.gov.au/Parliamentary_Business/Committees/House/Health_Aged_Care_and_Sport/Newdrugs/Report (viewed June 2022).
- Australian Government, Department of Health. Review of the National Medicines Policy [opened 30 Aug 2021; closed 8 Oct 2021]. https://consultations.health.gov.au/technology‐assessment‐access‐division/national‐medicines‐policy‐review/ (viewed June 2022).
Linked content
-
MJA Perspective: mRNA vaccines: a transformative technology with applications beyond COVID‐19
Provenance: Not commissioned; externally peer reviewed.