




Precision medicine aims to link molecular targets in cancers with corresponding therapies. A number of histotype-specific “basket trials” have yielded successful outcomes,1-4 employing genomic tools to identify potential therapeutic targets in cancer and test a range of targeted agents.
Molecular tumour profiling has now entered clinical practice, driven by capacity, reduced costs, and the enormous unmet need for effective treatments for advanced cancers.5 This particularly applies to rare cancers (defined by an incidence of less than 6 cases per 100 000 population), for which histotype-specific trials are rare. However, the translation of molecular profiling into clinical benefit requires testing in a clinical trial, and barriers to conducting such trials include complex regulatory processes, high costs, and long timelines.6-9
To translate molecular advances into clinical care for patients with rare cancers, innovative approaches are needed. The Cancer Molecular Screening and Therapeutics (MoST) program tests a novel paradigm for evaluating biomarker-driven treatments for patients with advanced cancer, with a particular focus on rare and neglected cancers. The overall aim is to develop a more effective infrastructure for testing novel rational therapeutic hypotheses. Additionally, we are undertaking longitudinal exploratory analyses of the understanding of and the attitudes to genomics among clinicians and patients.
Methods and analysis
Study design
The master protocol provides a structure for profiling tumours for actionable molecular targets and for evaluating treatments based on the molecular signatures of tumours, permitting the development and conduct of multiple, parallel, phase 1b–2a clinical substudies of novel treatments or indications for eligible patients. Actionable molecular targets are those clinically relevant to treatment. The recommended treatment can be a MoST substudy or require an alternative means of access to the relevant treatment. The master protocol specifies the design structure common to all clinical substudies, and each substudy is an addendum to the common design. The addenda provide sufficient details of each substudy to enable appropriate ethics and governance review, including of drug background and efficacy, rationale for molecular subtype selection, and substudy-specific eligibility criteria (example: Box 1).
Each substudy consists of one or more modules of 16 patients in an open label, single arm, signal-seeking design. A minimum of 12 substudy modules are planned for the first 4 years of the program. Substudies have one of three outcomes. First, negative results will be published and shared with the clinical community. Second, substudies finding a signal of efficacy may form the basis for an expanded substudy for investigating a refined version of the initial hypothesis and may lead to formal phase 2 testing. Third, for studies yielding an intermediate, promising result, but not sufficient for initiating a phase 2 trial, iterative substudies may be considered to increase the opportunity for detecting signals of drug activity. The MoST study also establishes the infrastructure for correlative, fundamental investigation of treatment response, tumour biology, and biomarkers, as well as for bioethical and psychosocial quantitative and qualitative assessments (study schema: Box 2).
Aims of the study
-
To identify signals of efficacy for developing biomarker-driven therapies;
-
To identify biomarkers that more accurately predict benefit of therapy;
-
To evaluate the utility of the MoST study design.
Study timetable and sites
The program commenced in October 2016 and will complete accrual in July 2019. Recruitment sites include the Kinghorn Cancer Centre at St Vincent’s Hospital Sydney, but additional sites will be added during 2018.
Target population
The target population comprises patients with pathologically confirmed advanced or metastatic solid cancers of any histological type, either during or after their last line of effective therapy. We are particularly interested in rare or neglected cancers and cancers of unknown primary site. Additional eligibility criteria are outlined in Box 3.
Study participation is divided into tumour molecular profiling and treatment phases, the latter typically based on the discovery of a suitable biomarker. Eligibility assessments and informed patient consent are obtained both prior to enrolment in the profiling phase and prior to the treatment phase. Patients deemed potentially eligible for treatment in a MoST substudy are assessed for eligibility according to criteria specific to the substudy (Box 3). Treatment may also be recommended outside MoST should there be a suitable clinical trial, or if an off-label therapy is appropriate.
Statistical aspects
A total of 1000 patients will be recruited during the molecular profiling phase, with the aim of identifying actionable mutations and determining eligibility for enrolment in MoST substudies. It is planned that a minimum of 12 substudy modules will be opened in the initial year of the program, each enrolling 16 patients. As the therapeutic substudies are signal-seeking in nature and involve a heterogeneous group of tumour histologies, formal power calculations cannot be undertaken. A module size of 16 was chosen as sufficient for detecting a signal of therapeutic efficacy, in analogy to the first of the two stages of the Simon phase 2 trial design, in which a first signal-seeking phase with 10–16 participants is typical when determining whether formal expansion into a larger phase 2 trial is justifiable.10 A primarily descriptive analysis will evaluate the feasibility, efficiency, utility, and costs of the master protocol, with data on the development and conduct of each substudy contributing to this analysis.
In diseases that are refractory to treatment, a response in at least three of 16 patients is regarded as clinically meaningful, justifying further clinical evaluation; substudies with fewer responders are generally viewed as not supporting the molecular hypothesis underlying the substudy.11 If appropriate, iterative substudies may be considered to increase the opportunity for detecting signals of drug activity.
Interventions: molecular profiling phase
Retrieval of archival biospecimens
Archival formalin-fixed, paraffin-embedded diagnostic material sourced from pathology laboratories is used for molecular profiling. In addition, a blood sample is obtained for subtractive analysis of somatic mutation status if required, and for future (unspecified) research. Details about collecting biomarker and future research samples for companion studies will be specified in each substudy addendum.
Molecular profiling and other molecular assays
All biospecimen collection and processing is conducted in accordance with the Australasian Biospecimen Network Biorepository Protocols.12 The molecular profiling assay is based on genomic sequencing for a broad range of biologically important cancer genes. Our current protocol employs the TruSight Tumor 170 panel (Illumina), which includes both DNA and RNA components. These assays identify single nucleotide variations, insertions, deletions, highly amplified genes, and gene fusions. The sequencing panel is updated iteratively throughout the program to include new targets, and will be improved to reflect the evolution of the field and for developing new substudies. Patient tumour samples may also be assessed for biomarkers using other assays (immunohistochemistry, fluorescence in situ hybridisation) according to new therapeutic hypotheses. Validation of the identified targets will be performed with an appropriate orthogonal technique, a test accredited by the National Association of Testing Authorities (https://www.nata.com.au), or a trial-specific assay.
Molecular review and variant classification
The outputs of tumour genomic sequencing are subjected to bioinformatics analysis and reviewed by the Garvan Molecular Tumour Board (MTB) for variant classification and assignment of “actionability” category on the basis of information in the published literature, patient history, and availability of therapeutic opportunities.
There are three categories of actionable variants:
-
mutations that mean the patient is suitable for a MoST substudy;
-
mutations for which an existing funded or unfunded drug is available; and
-
mutations that mean the patient is suitable for an existing clinical trial.
If several molecular targets are found, the MTB will make a recommendation based on the clinical profile of the patient and expected pathogenicity of each mutation; that is, its importance in the context of other mutations, and its role as a driver of tumour growth and progression.
Informing patients about molecular profiling results
All participants, including those with no actionable biomarkers, are informed of their tumour profiling results. The MTB report is provided to the patient’s clinicians for discussion with the patient. The decision to participate in a MoST substudy is made by the patient in consultation with their referring oncologist.
Ethically defensible plan for disclosing clinically significant information
Clinically significant cancer-related heritable genetic factors with implications for future health are sometimes identified. Research findings are reviewed by the MTB and other relevant expert committees to determine clinical significance in accordance with national and international guidelines.13-16 During the consent process, participants are asked whether they wish to receive information on hereditary cancer risks of potential importance to their future health or that of their blood relatives. Participants indicating that they wish to be informed are notified in writing that a mutation has been found, and are invited to contact a genetic counsellor, who may in turn arrange contact with a familial cancer clinic or other medical professional, as appropriate.
Interventions: treatment phase
Two general classes of drugs will be used in MoST substudies:
-
drugs approved for clinical use in Australia by the Therapeutic Goods Administration (TGA), but not for the indication being treated;
-
drugs not approved for clinical use in Australia by the TGA, but which are being tested in phase 2 or 3 clinical trials for other indications, or are registered outside Australia.
The drugs employed will have well characterised toxicity profiles and established dose and administration schedules.
Outcomes
Overall outcome measures include assessment of the trial design, molecular profiling, and individual outcomes from substudies. A major technical problem has been developing broadly applicable outcome measures because of the diversity of cancer types included in MoST.
Measures for evaluating the modular trial design
-
Key milestones, including the length of time:
-
from proposal to opening of a MoST substudy;
-
from ethics approval for the MoST program to opening of the first substudy;
-
from ethics approval for the addenda to the opening of substudies;
-
from completion of enrolment to study closure; and
-
from study closure to reporting of results.
-
-
The number of MoST substudies that yield a positive signal of activity and proceed to formal phase 2 testing.
-
The number of biomarkers of response that are identified.
-
Data from unsuccessful substudies (trial concepts that do not progress to a substudy in the MoST program).
Measures for evaluating the molecular screening platform
-
The number of actionable molecular biomarkers identified by tumour profiling.
-
The number of patients whose therapy is altered as the result of molecular tumour profiling.
-
The number of patients enrolled in screening over defined periods.
-
The number of patients eligible for enrolment and registered in a MoST substudy.
-
The time from patient consent to communication of profiling results to the patient’s clinician (date of issue of the MTB report).
-
The time from identification of an actionable mutation by the MTB to registration of the patient in a substudy.
-
Qualitative and quantitative analyses of the molecular profiling assays, including sequencing failure rate.
-
Overall survival of patients whose treatment was altered as a result of molecular profiling, compared with that of patients who received standard therapy.
-
Qualitative and quantitative analyses of patients’ expectations and experiences of genomic screening and of clinical outcomes.
Treatment phase: measures for evaluating clinical activity of treatments tested in substudies
The primary endpoint for assessing efficacy and decision making in all substudies will be disease control, defined as the proportion of patients for whom:
-
an objective tumour response is observed: complete and partial responses according to cancer-specific response criteria; or
-
time to progressive disease during the substudy (TTP2) exceeds the documented time to progressive disease during the last treatment prior to substudy entry (TTP1) by at least 30% (ie, TTP2/TTP1 ≥ 1.3),4 or, if TTP1 is not evaluable, time to progressive disease exceeds 6 months.
Response status (ie, complete response, partial response, stable disease, progressive disease) will be determined by conventional criteria.17,18
Stabilisation of disease is also taken into account in the TTP2/TTP1 component of the composite primary endpoint. Given the limited historical data available for rare cancer types and the heterogeneity of the population studied, it is not possible to define a meaningful minimal interval during which stable disease criteria should be maintained. In the TTP2/TTP1 ratio, each patient acts as their own control, and patients who achieve stable disease relevant to disease control are included. This methodology has been applied in previous basket studies.4 When TTP1 is not available, the minimum period of time for stable disease is set at 6 months.
Due to the heterogeneity of tumour types, it may be necessary to define a range of specific measures for determining treatment response in some patients. In those with specific cancer subtypes, biomarker-based or qualitative clinical assessment of response according to defined criteria may be included. Alternative validated guidelines are employed when response cannot be evaluated with Response Evaluation Criteria in Solid Tumours (RECIST) 1.1.17
The secondary endpoints are defined as:
-
overall survival: the interval from the date of registration to date of death from any cause or to the date of last known follow-up;
-
safety and tolerability of treatment: rates of adverse events graded with the National Cancer Institute Common Terminology Criteria for Adverse Events 4.03 (NCI CTCAE);19
-
health-related quality of life during substudy treatment: assessed with the EORTC QLQ-C30 (version 3) questionnaire20 or Brief Pain Inventory21 as applicable.
Additional measures relevant to the assessment of clinical or biological activity of the study treatment collected as secondary outcomes include associations of response, resistance, or tolerability with biomarkers. The precise nature of any additional data to be collected is specified in each substudy addendum.
Bioethical and psychosocial quantitative and qualitative assessments
Validated patient-reported outcomes for assessing the patient’s knowledge, values, attitudes, coping strategies, and decisional and psychosocial outcomes are collected during consent to molecular profiling, immediately after return of profiling results, and 2 months later. At these assessment points, patients are invited to participate in an audiotaped interview to explore these questions in greater depth.
Health-related quality of life
Health-related quality of life is measured by performance status assessments and with the EORTC QLQ-C30 questionnaire20 during the treatment phase. The severity of pain and of pain associated with daily functions is assessed with the Brief Pain Inventory short form when appropriate.21
Follow-up
Molecular profiling
Clinical data are collected for all patients, including those not eligible for participation in a clinical trial substudy. All patients are followed up at 3, 6, and 12 months and at 2 years after entry into the molecular profiling phase to determine survival and disease status and whether or not they received matched molecular therapy on the basis of tumour profiling (Box 4).
Substudy
Patients enrolled in a substudy are followed up every 4 weeks until progression, or until end of treatment and for at least 30 days after the end of treatment. The date of death is ascertained in medical records and appropriate registries.
Ethics approval
The framework protocol and each substudy addendum have been reviewed in full and approved by the St Vincent’s Hospital Sydney Human Research Ethics Committee (reference, HREC/16/SVH/23).
Funding statement
MoST is funded by the New South Wales Office for Health and Medical Research (reference, H15/13965; 2 March 2015). Therapeutic substudies undertaken to date have been funded and drugs supplied by AstraZeneca Australia and Pfizer. The psychosocial substudy is funded by a National Health and Medical Research Council project grant (APP1124749).
Dissemination of findings
Our results will be disseminated in the clinical study report, presented at national and international conferences and scientific meetings, and published (as aggregated data not identifying individual participants) in peer-reviewed journals. All study data will be stored in a secure area or on secure servers accessible only to authorised staff members.
Author contributions
Subotheni Thavaneswaran implemented and critically evaluated the protocol, refined data collection and information dissemination to referring oncologists and patients, and reviewed and approved the final manuscript.
Lucille Sebastian drafted and critically reviewed the protocol, implemented the design and conduct of substudies, including planning and execution of substudies and data collection, and reviewed and approved the final manuscript.
Mandy Ballinger implemented and critically evaluated the protocol, refined data collection and information dissemination, and reviewed and approved the final manuscript.
Megan Best contributed to the psychosocial substudy (longitudinal survey and interview).
Dominique Hess drafted and critically reviewed the protocol, implemented the design and conduct of screening infrastructure, and reviewed and approved the final manuscript.
Chee Lee critically reviewed the protocol, supervised the design and conduct of substudies, and reviewed and approved the final manuscript.
Katrin Sjoquist critically reviewed the protocol, supervised the design and conduct of substudies, and reviewed and approved the final manuscript.
Wendy E Hague critically reviewed the protocol, supervised the design and conduct of substudies, and reviewed and approved the final manuscript.
Phyllis N Butow contributed to the psychosocial substudy (longitudinal survey and interview).
John Simes drafted and critically reviewed the protocol, supervised the design and conduct of substudies, and reviewed and approved the final manuscript.
David Thomas conceptualised the design, drafted and critically reviewed the protocol, supervised the conduct of screening and substudies, and reviewed and approved the final manuscript.
Contributions by non-authors
Anthony Joshua, Amy Prawira (Kinghorn Cancer Centre, St Vincent's Hospital Sydney): clinical implementation of the framework and substudy protocols
Min Ru Qiu (SydPath, Sydney): quality assessment of archival material.
Elektra Hajdu, Audrey Silvestri (Garvan Institute of Medical Research, Sydney): biospecimen processing and molecular screening of archival tumour material; orthogonal validation of screening results.
John Grady, Mark Cowley, Mark Pinese (Garvan Institute of Medical Research, Sydney): bioinformatic analyses.
Gary Pan, Emily Collignon, Kelly Prendergast, Christine Napier (Garvan Institute of Medical Research, Sydney): study and participant coordination.
Michael Tayao, Andrew Da Silva, Leeda Zamiri, Anaiis Zaratzian (Garvan Institute of Medical Research, Sydney): biospecimen processing and storage.
Box 1 – The palbociclib substudy as an example of the concepts outlined in the protocol and its relationship to therapeutic substudies
- The retinoblastoma protein (pRb)–cell cycle pathway is critical to cancer development. Mutations in genes coding for components of this pathway, including the cyclin-dependent kinases CDK4, CDK6 and CDKN2A, and cyclin D1, are frequent in people with cancer. The drug palbociclib targets a central component of this pathway, CDK4.
- We hypothesised that patients whose tumours contain mutations in CDK4 pathway genes might benefit from treatment with palbociclib. The MoST study has identified 39 patients (among 476 patients, 8%) who carry eligible mutations in CDK4 pathway genes. Sixteen people with four cancer types, including six sarcoma subtypes, were enrolled in the palbociclib substudy between November 2016 and December 2017.
- Depending on the final outcomes, we may:
- consider extending the substudy to include an additional 16 participants;
- conclude that the evidence for the effectiveness of palbociclib is insufficient to justify further exploration; or
- develop a proposal for a formal phase 2 clinical trial in this population for evaluating its efficacy.
- This substudy has entered the follow-up period; we anticipate reporting our findings by late 2018.
Box 2 – Framework protocol of the Cancer Molecular Screening and Therapeutics (MoST) program
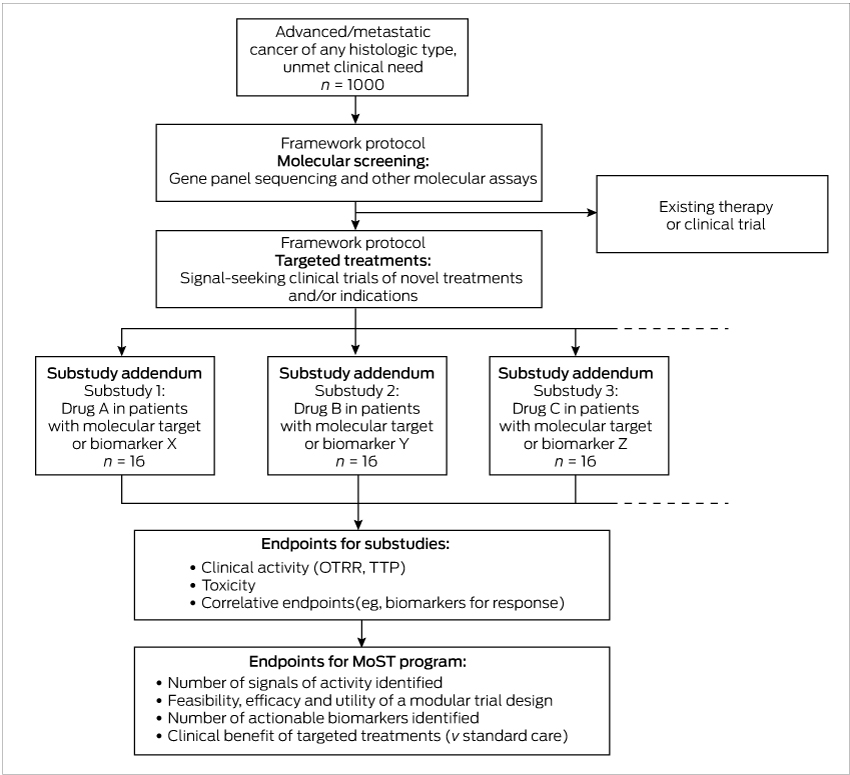
OTRR = objective tumour response rate; TTP = time to progression.
Box 3 – Inclusion and exclusion criteria for the molecular profiling and substudy treatment phases of the Cancer Molecular Screening and Therapeutics (MoST) program
|
Molecular profiling phase: inclusion criteria |
Molecular profiling phase: exclusion criteria |
||||||||||||||
|
|
|||||||||||||||
|
|
|
||||||||||||||
|
Treatment phase: inclusion criteria |
Treatment phase: exclusion criteria |
||||||||||||||
|
|
|
||||||||||||||
|
|
|||||||||||||||
|
ALT = alanine transaminase; AST = aspartate aminotransferase; ECOG = Eastern Cooperative Oncology Group; ULN = upper limit of normal. |
|||||||||||||||
Box 4 – Schedule of assessments for the Cancer Molecular Screening and Therapeutics (MoST) program
|
|
Screening |
Baseline |
During treatment |
Follow-up after end of treatment |
|||||||||||
|
During 14 days before registration |
During 7 days before registration |
Each cycle, before treatment |
8-weekly during treatment |
Other |
30 days after last dose |
Every 4 weeks until progression |
|||||||||
|
|
|||||||||||||||
|
Confirmation report from MTB |
X |
|
|
|
|
|
|
||||||||
|
Informed consent |
X |
|
|
|
|
|
|
||||||||
|
Clinical assessment |
|
X |
X |
|
|
X |
|
||||||||
|
Haematology* |
|
X |
X |
|
|
|
|
||||||||
|
Biochemistry* |
|
X |
X |
|
|
|
|
||||||||
|
Concomitant medications report |
|
|
|
|
During an SAE |
|
|
||||||||
|
Imaging (eg, CT, MRI, PET) |
X† |
|
X |
|
|
X‡ |
|||||||||
|
Circulating biomarkers |
X |
|
X |
X |
X |
|
|
||||||||
|
Pregnancy test |
|
X |
|
|
|
|
|
||||||||
|
Adverse events |
|
|
X |
|
|
X |
|
||||||||
|
Biospecimen collection |
|
As specified in each substudy addendum |
|
|
|||||||||||
|
Quality of life assessment |
|
|
X |
|
|
|
X |
||||||||
|
Pain scale |
|
X |
X |
|
|
|
X |
||||||||
|
|
|||||||||||||||
|
CT = computerised tomography; MRI = magnetic resonance imaging; MTB = Molecular Tumour Board; PET = positron emission tomography; SAE = serious adverse event. * If sample collected within 48 hours of each time point (eg, treatment), assessment does not need to be repeated, including baseline. † Imaging should be undertaken in the 21 days preceding registration. ‡ Imaging continues on an 8-weekly schedule after the end of treatment. |
|||||||||||||||
- Subotheni Thavaneswaran1,2
- Lucille Sebastian3
- Mandy Ballinger1
- Megan Best4
- Dominique Hess1
- Chee K Lee3
- Katrin M Sjoquist3
- Wendy E Hague3
- Phyllis N Butow5
- R John Simes3
- David Thomas1,2
- 1 Garvan Institute of Medical Research, Sydney, NSW
- 2 St Vincent's Clinical School, University of New South Wales, Sydney, NSW
- 3 NHMRC Clinical Trials Centre, University of Sydney, Sydney, NSW
- 4 Psycho-Oncology Co-operative Research Group (PoCoG), University of Sydney, Sydney, NSW
- 5 Centre for Medical Psychology and Evidence-based Decision Making, University of Sydney, Sydney, NSW
No relevant disclosures.
- 1. Andre F, Bachelot T, Commo F, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol 2014; 15: 267-274.
- 2. Arnedos M, Scott V, Job B, et al. Array CGH and PIK3CA/AKT1 mutations to drive patients to specific targeted agents: a clinical experience in 108 patients with metastatic breast cancer. Eur J Cancer 2012; 48: 2293-2299.
- 3. Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res 2012; 18: 6373-6383.
- 4. Von Hoff DD, Stephenson JJ, Rosen P, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010; 28: 4877-4883.
- 5. Ward R. A decade of promises in personalised cancer medicine: is the honeymoon over? Med J Aust 2014; 200: 132-133. <MJA full text>
- 6. Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov 2012; 11: 191-200.
- 7. Light DW, Kantarjian H. Market spiral pricing of cancer drugs. Cancer 2013; 119: 3900-3902.
- 8. Steensma DP, Kantarjian HM. Impact of cancer research bureaucracy on innovation, costs, and patient care. J Clin Oncol 2014; 32: 376-378.
- 9. Stewart DJ, Whitney SN, Kurzrock R. Equipoise lost: ethics, costs, and the regulation of cancer clinical research. J Clin Oncol 2010; 28: 2925-2935.
- 10. Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials 1989; 10: 1-10.
- 11. Mehta CR, Cain KC. Charts for the early stopping of pilot studies. J Clin Oncol 1984; 2: 676-682.
- 12. Mallette A, Tassé AM. P3G model framework for biobank access policy: core elements. Dec 2013. http://p3g2.org/wp-content/uploads/P3G-Core-Elements-Access-Policy-Final-Dec-2013-1.pdf (viewed Aug 2018).
- 13. Green RC, Berg JS, Grody WW, et al; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013; 15: 565-574.
- 14. Kalia, SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017; 2: 249-255.
- 15. Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405-424.
- 16. Cancer Institute NSW. Cancer genetics. eviQ Cancer Treatments Online [website]. 2017. https://www.eviq.org.au/cancer-genetics (viewed July 2018).
- 17. Eisenhauer E, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228-247.
- 18. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 2010; 28: 1963-1972.
- 19. National Cancer Institute (United States). CTCAE files. https://evs.nci.nih.gov/ftp1/CTCAE/About.html (viewed July 2018).
- 20. Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst 1993; 85: 365-376.
- 21. Cleeland CS, Ryan KM. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singapore 1994; 23: 129-138.
Abstract
Background: Precision medicine aims to link molecular targets in tumours with corresponding therapies, particularly for patients with rare cancers. Innovative approaches are needed to translate molecular opportunities into clinical care. The Cancer Molecular Screening and Therapeutics (MoST) program employs a molecular screening platform to identify molecular changes of therapeutic relevance (actionable changes) and a master protocol for multiple, parallel signal-seeking clinical substudies, focused on therapies for patients with rare and neglected cancers.
Methods and analysis: Archival pathology laboratory samples from patients with treatment-refractory advanced solid cancer of any histologic type undergo molecular tumour profiling. Following review by a Molecular Tumour Board, eligible patients are offered treatment in therapeutic substudies. This novel master protocol allows expedited addition of individual substudies; at least 12 open label, single arm, signal-seeking substudies during the initial 4 years of MoST are planned. The primary objectives are to identify signals of efficacy for developing biomarker-driven therapies and biomarkers that more accurately predict response to therapy, as well as to evaluate the MoST design.
Ethics approval: The program has been approved by the St Vincent’s Hospital Sydney Human Research Ethics Committee (reference, HREC/16/SVH/23).
Dissemination of results: A report summarising and interpreting collected study data will be published. Our findings will be presented at national and international conferences and scientific meetings, and published in peer-reviewed journals.
Trial registration: Australia New Zealand Clinical Trials Registry: ACTRN12616000908437 (8 July 2016).