





Volume 206 - Issue 7
Translating our microbiome into medicine
Authors: Mark Morrison and Gerald Holtmann
Med J Aust 2017; 206 (7): 287-288. || doi: 10.5694/mja17.00087
Published online: 17 April 2017
Published online: 17 April 2017
Integrating contemporary microbiology with new sequencing technologies will allow us to better understand our microbiome and its relationships with health and disease
Integrating contemporary microbiology with new sequencing technologies will allow us to better understand our microbiome and its relationships with health and disease
We live in a microbial world. We are surrounded by and in contact with microbes that support many natural and managed processes, from carbon capture by microalgae to support marine food webs, to the fermentations that produce beverages and foods that we consume and enjoy. Nor is our body a sterile environment, either inside or out. While it has long been recognised that animals, including humans, are colonised soon after birth (or hatching), changes in our perceptions of the human microbiota have arisen over the past decade via the step advances made in DNA and RNA amplification methods, sequencing technologies and computational biology — what has been referred as the “omics” era of research. All these approaches support an assessment of the microbial world without first having to culture microbes in a laboratory setting, and the capacity to characterise both the structure and function of entire microbial communities from real-world samples.
During the past decade, initiatives in North America, Europe and China have been at the forefront of providing a genomic-based characterisation of our microbiota — this is generically referred to as our microbiome, with the term “microbiota” restricted to the viable state.1 If our body is considered as a landscape, then it possess multiple environments with variations in temperature, water content, pH, exposure to light and gases, and oxygen availability. Despite these variations, microbiome profiling studies using 16S ribosomal RNA gene sequencing2 have shown that our recruitment of the microbial world to reside and interact with us is rather selective, constrained to around 10 of the 50 or so phyla that are currently known to exist on our planet, and with each site possessing its own signature profile of members from this broadest taxonomic classification.3 Evidence also now suggests that our first encounter with the microbial world occurs in utero via a placental microbiome,4 and other body sites and organs once considered to be sterile under healthy conditions, such as the lung, are now considered to possess their own community of commensal microorganisms.5 Even body sites such as the oral cavity and large bowel, which have long been studied using classical microbiology techniques, have been transformed in terms of our understanding of their form and function. We have learned that our microbiota ages with us, and can be shaped in a deterministic way by our own genetics and dietary pattern.6 More recently, time and cost constraints have lessened, allowing metagenomic sequencing,2 which provides an inventory of the complete genetic potential inherent to microbial communities, and may soon supplant microbiota profiling approaches. Both approaches have further expanded our awareness of the scope of the nutritional, structural and physiological impacts that the gut microbiota may have on host metabolism, (immuno)physiology and other homeostatic processes. Indeed, observational, cross-sectional and case–control studies have now been published reporting changes in the microbiome that are associated with various medical conditions. This has led to the use of the term “dysbiosis” to refer collectively to what is considered an aberrant microbial profile present with these conditions when compared with healthy subjects.7
In a study of the duodenal mucosal microbiome, patients with functional dyspepsia were found to have a greater relative abundance of Streptococcus and decreases in the relative abundance of other genera such as Prevotella, Veillonella and Actinomyces compared with control subjects, suggesting that their symptoms may be related to alterations of their microbiome at this site (Box).8
It has also long been recognised that some animal models of disease, when maintained in a germ-free state, remain free of the phenotype. For example, germ-free animal models of inflammatory bowel disease remain quiescent until microbiota are introduced, which results in the rapid onset of disease.9 Conceptually similar observations have recently been made with respect to the role of the gut microbiota in trimethylamine formation from choline-rich diets and cardiovascular disease pathogenesis.10 Gnotobiotic mouse models are also now being used to validate that some specific commensal gut bacteria have protective effects, by positively affecting gut homoeostasis and attenuating inflammation or disease. For instance, Miquel and colleagues11 used gnotobiotic mice (Escherichia coli ± Faecalibacterium prausnitzii) to show that if F. prausnitzii is maintained at high levels in colitic mice, the disruption of the colonic epithelium is greatly attenuated.
Additionally, microbiota transplants in mice, using faecal slurries prepared from humans or from mice with specific phenotypes, have become a popular route for establishing that the microbiota, as a community, can elicit phenotypic changes in the host, from obesity12 to depressive-like behaviours.13 Such findings, along with the now well established clinical efficacy of faecal microbiota transplants (FMT) for alleviating Clostridium difficile infections, have led to a resurgent interest in this approach in Australia14 and abroad to treat a variety of medical conditions where gut dysbiosis is implicated.15,16 However, the current optimism for the use of FMT to treat a broader range of digestive diseases is moderated by our current understanding of the microbiome from both a donor and recipient context. While the donor’s stool microbiome (or its preparation) is widely accepted as a key influence on the therapeutic effect, it is also possible that patients could be exposed to additional risks transmitted via a donor’s stool. The resilience of the recipient’s microbiome to perturbations like FMT, and its recovery from such a challenge, will also likely affect efficacy. In that context, our gut microbiota is considered to have developed resilience relatively early in life,6 but our understanding of the concept of resilience and strategies that might support our management of it in a clinical setting are largely unexplored. Interestingly, a recent report of vaginal microbiota transfer from mothers to their infants delivered by caesarean section suggests this intervention, at a time when the human microbiota is most dynamic and pliable, might promote the development of a skin and oral microbiota more similar to that of their mothers compared with untreated infants.17 In summation, while the findings above represent some of the many findings linking our microbiota with our health, and reveal much promise and optimism for the use of our microbiota for future treatments, a combination of caution and refinement of approaches remains vital from an ethical, legal and social context. To this end, efforts such as the European consensus conference on FMT in clinical practice18 seek to promote an evidence-based approach as guidance for interested physicians.
In relation to key knowledge gaps that constrain the translation of our microbiome into medicine, metagenomic analysis of stool microbiota from various origins (Europe, North America, and Japan) suggests that these communities are similar but not universal.19 To date, equivalent datasets have not been produced more broadly across the Asia–Pacific region, despite the region’s genetically diverse populations with vastly different diets and lifestyles compared with those of the Americas, Europe, Africa and China. Common to these ethnic groups are the burdens of many, but not all, chronic Western immune-mediated and metabolic diseases and cancers, and at rapidly increasing incidence rates, suggesting a non-genetic basis for their penetration into these communities. Moreover, while the omics-based approaches have produced substantial advances in our knowledge of the human microbiome, much remains as “dark matter” in the form of unclassified genes or metagenomic species assembled from the data, including viruses, but for which no microbial isolate or biochemical characterisation is available.20 Indeed, we contend that there has been, to some extent, the development of a culture gap, both literally and figuratively, between the genomics-driven approaches and the other research elements inherent to environmental microbiology, which needs to be corrected. In this context, the lower eukaryotes (protozoa, yeasts and fungi) have been somewhat neglected during the omics era — in particular their roles as members of the gut microbiota21 — and warrant further investigation. It should also be noted that most research to date has focused on the stool microbiome. However, colonisation of the mucosa by specific bacteria (the mucosa-associated microbiome) and/or the study of microbiomes resident within other surfaces of the human body might be more relevant with respect to other medical conditions afflicting different body sites and organs.
In conclusion, exploring the microbiome and, in particular, the interactions between the microbiome and the human host, may substantially add to our knowledge of the pathophysiology of many diseases. While targeting the microbiome may offer new approaches to prevent or treat a broad spectrum of diseases, the interactions between host and the microbiome are complex. Simple but crude measures such as exposing patients to stool preparations (FMT) from healthy subjects might be attractive, but in the long term are unlikely to be the way forward, considering safety aspects and the need for standardised and contamination-free microbiota preparations that comply with good manufacturing practice standards. On the other hand, modulation of the microbiome with currently available probiotics — although more likely to be compliant with relevant manufacturing standards — may not deliver the potential benefits. Omics-based approaches will be crucial to revealing potential roles for the microbiome in health and disease, and when combined with the principles of environmental microbiology, including culture, offer the opportunity to shine a light on our microbial dark matter and refine the concepts of probiotics, prebiotics and FMT. The continued support and inspiration of the medical community will therefore be key for catalysing the translation of our new understanding of the microbiome into medicine.
Box – Difference in the duodenal microbiome of patients with functional dyspepsia (FD) compared with controls, using 16S ribosomal RNA gene sequencing
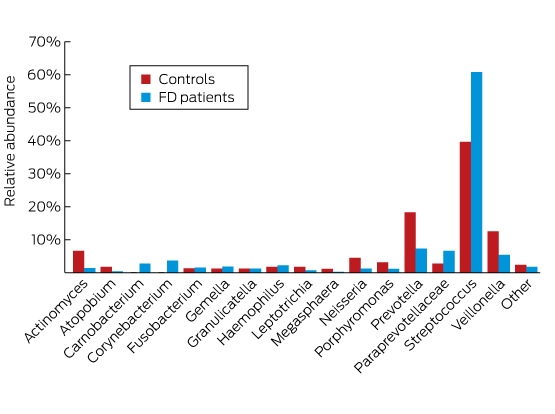
Adapted with permission from Zhong et al.8
Competing interests
No relevant disclosures.
References
- Hanson BM, Weinstock GM. The importance of the microbiome in epidemiologic research. Ann Epidemiol 2016; 26: 301-305.
- Morgan XC, Huttenhower C. Chapter 12: Human microbiome analysis. PLoS Comput Biol 2012; 8: e1002808.
- Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 2011; 9: 279-290.
- Collado MC, Rautava S, Aakko J, et al. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep 2016; 6: 23129.
- Budden KF, Gellatly SL, Wood DL, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol 2017; 15: 55-63.
- Ottman N, Smidt H, de Vos WM, et al. The function of our microbiota: who is out there and what do they do? Front Cell Infect Microbiol 2012; 2: 104.
- Harmsen HJ, de Goffau MC. The human gut microbiota. Adv Exp Med Biol 2016; 902: 95-108.
- Zhong L, Shanahan ER, Raj A, et al. Dyspepsia and the microbiome: time to focus on the small intestine. Gut 2016; doi: 10.1136/gutjnl-2016-312574 [Epub ahead of print].
- Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 1998; 66: 5224-5231.
- Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011; 472: 57-63.
- Miquel S, Leclerc M, Martin R, et al. Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. mBio 2015; 6: e00300-e00315.
- Graham C, Mullen A, Whelan K. Obesity and the gastrointestinal microbiota: a review of associations and mechanisms. Nutr Rev 2015; 73: 376-385.
- Zheng P, Zeng B, Zhou C, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatry 2016; 21: 786-796.
- Costello SP, Tucker EC, La Brooy J, et al. Establishing a fecal microbiota transplant service for the treatment of Clostridium difficile infection. Clin Infect Dis 2016; 62: 908-914.
- Borody T, Fischer M, Mitchell S, et al. Fecal microbiota transplantation in gastrointestinal disease: 2015 update and the road ahead. Expert Rev Gastroenterol Hepatol 2015; 9: 1379-1391.
- Paramsothy S, Walsh AJ, Borody T, et al. Gastroenterologist perceptions of faecal microbiota transplantation. World J Gastroenterol 2015; 21: 10907-10914.
- Dominguez-Bello MG, De Jesus-Laboy KM, Shen N, et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med 2016; 22: 250-253.
- Cammarota G, Ianiro G, Tilg H, et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017; doi: 10.1136/gutjnl-2016-313017 [Epub ahead of print].
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464: 59-65.
- Nielsen HB, Almeida M, Juncker AS, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol 2014; 32: 822-828.
- Liguori G, Lamas B, Richard ML, et al. Fungal dysbiosis in mucosa-associated microbiota of Crohn’s disease patients. J Crohns Colitis 2016; 10: 296-305.
Linked content
-
MJA InSight: Translating our microbiome
Provenance: <p>Commissioned; externally peer reviewed.</p>