




An update on research advances in this field that may help tackle this growing challenge more effectively
Increasing life expectancy has fuelled the growth in the prevalence of dementia. In 2015, there were an estimated 47 million people with dementia worldwide (including 343 000 in Australia), a number that will double every 20 years to 131 million by 2050 (900 000 in Australia).1 The global cost of dementia in 2015 was estimated to be US$818 billion.1 Low-to-middle income countries will experience the greatest rate of population ageing, and the disproportionate growth in dementia cases in these nations will be exacerbated by a relative lack of resources.
The diagnostic criteria for dementia (relabelled “major neurocognitive disorder”) of the American Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5)2 include a significant decline in one or more cognitive domains that is clinically evident, that interferes with independence in everyday activities, and is not caused by delirium or other mental illness. Whether the new diagnostic label catches on remains to be seen. The most common type of dementia is Alzheimer’s disease (AD) (50–70% of patients with dementia), followed by vascular dementia (10–20%), dementia with Lewy bodies (10%) and fronto-temporal dementia (4%).3 These percentages are imprecise, as patients often present with mixed pathology.
Our discussion will focus on AD because it receives significant research attention as the most common cause of dementia. The two hallmark pathological changes associated with neuronal death in AD are deposition of β-amyloid plaques, and tau protein neurofibrillary tangles. Understanding this process has been enhanced by prospective cohort studies, such as the Australian Imaging Biomarkers and Lifestyle (AIBL) study.4 As shown in the Box, the results of this research indicate that the degree of β-amyloid deposition exceeds a predefined threshold about 17 years before the symptoms of dementia are detectable. In the absence of an alternative model, the amyloid cascade remains the most compelling hypothesis for the pathogenesis of AD. This is supported by the fact that early onset familial AD is caused by mutations in chromosome 21 that result in the production of abnormal amyloid precursor protein (APP), or by mutations in chromosomes 1 or 14 that result in abnormal presenilin, each of which increase amyloid deposition. The extra copy of chromosome 21 in Down syndrome also leads to faster amyloid deposition and the earlier onset of AD. Further, the symptoms of AD are correlated with imaging of amyloid in the living brain and with cerebrospinal fluid biomarkers that are now included in new diagnostic criteria for AD and which will enable suitable participants to be selected for trials of drugs that may prevent or modify the disease,2 in particular to determine whether anti-amyloid agents are useful for delaying or treating AD.
At present, cholinesterase inhibitors (donepezil, galantamine and rivastigmine) and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine are licensed for treating AD dementia, and produce modest but measurable benefits for some patients. These medications are thought to work by increasing cholinergic signalling and reducing glutamatergic activity respectively, partially redressing neurochemical abnormalities caused by the amyloid cascade.5 More than 200 other drugs advanced to at least Phase II development between 1984 and 2014, but none has yet entered routine clinical use.6 Lack of efficacy in clinical trials may be the result of their being introduced at a rather late stage of the disease process; hippocampal damage is so profound by the time individuals present with AD dementia that attempting to slow their decline with an anti-amyloid agent may be analogous to starting statins in patients on a heart transplantation waiting list. As it provides the most compelling hypothesis for AD, the amyloid cascade remains the main target for developments in treatment. Treatment trials in people with preclinical or prodromal AD will in due course determine its validity.
Recent developments include promising results for treating prodromal AD with passive vaccines containing monoclonal antibodies directed against β-amyloid, such as solanezumab and aducanumab. This may point the way to treatments in the next decade that delay the onset of dementia in people with developing AD pathology.7,8
The identification of risk factors for AD may lead to risk reduction strategies. Recent randomised controlled trials of multidomain interventions, such as the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER) study (a 2-year program including dietary, exercise, cognitive training and vascular risk monitoring components), show that such interventions could improve or maintain cognition in at-risk older people in the general population.9 Greater risk reduction might be attained by intervening 10 to 20 years before the first clinical signs of cognitive impairment are presented. A recent review of 25 risk and protective factors associated with AD concluded that “the evidence is now strong enough to support personalized recommendations for risk reduction by increasing levels of education in young adulthood, increasing physical, cognitive and social activity throughout adulthood, reducing cardiovascular risk factors including diabetes in middle-age, through lifestyle and medication, treating depression, adopting a healthy diet and physical activity, avoiding pesticides and heavy air pollution and teaching avoidance of all potential dangers to brain health while enhancing potential protective factors”.10 These risk factors, and particularly vascular risk factors, are implicated in neurodegeneration pathology in a number of dementia processes.
While the search for effective preventive strategies and access to evidence-based pharmacological treatments and psychosocial interventions are critical, there are still delays in diagnosis and a failure to utilise existing available resources.1,3 The introduction of the federal government-funded, state-based Dementia Behaviour Management Advisory Services (DBMAS), the initiation of severe behaviour response teams, and increased funding for research should be applauded, but there needs to be greater coordination of service delivery systems for patients and carers at every stage, from prevention through to end-of-life care, and the medical profession needs to do more to ensure that all existing and trainee practitioners are well informed about what we can do for people with dementia right now.
Box – Relationship of ß-amyloid deposition with other parameters in Alzheimer disease
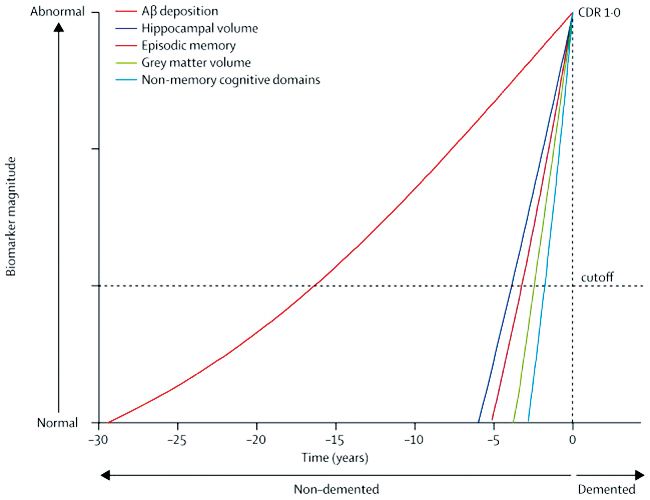
Aß-amyloid = ß-amyloid; CDR = Clinical Dementia Rating. Reproduced with permission from Villemagne et al (2013).4
Provenance: Commissioned; externally peer reviewed.
- 1. Prince M, Wimo A, Guerchet M, et al. World Alzheimer report 2015. The global impact of dementia: an analysis of incidence, prevalence, cost and trends. London: Alzheimer’s Disease International 2015. http://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf (accessed Sept 2015).
- 2. American Psychiatric Association: Diagnostic and statistical manual of mental disorders, 5th ed. Arlington: APA, 2013.
- 3. Ames D, Burns A, O’Brien J. Dementia, 4th ed. London: Hodder Arnold, 2010.
- 4. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid ß deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol 2013; 12: 357-367.
- 5. Parsons CG, Danysz W, Dekundy A, Pulte I. Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox Res 2013; 24: 358-369.
- 6. Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med 2014; 275: 251-283.
- 7. Sevigny J, Chiao P, Williams L, et al. Aducanumab (BIIB037), an anti-amyloid beta monoclonal antibody, in patients with prodromal or mild Alzheimer’s disease: interim results of a randomized, double-blind, placebo-controlled, phase 1b study [abstract: Alzheimer’s Association International Conference; 2015 July 18-23; Washington DC, USA]. Alzheimers Dement 2015; 11 Suppl: P277.
- 8. Liu-Seifert H, Aisen PS, Andersen SW, et al. Delayed-start analyses of up to 3.5 years in the phase 3 solanezumab expedition program in mild Alzheimer’s disease [abstract: Alzheimer’s Association International Conference; 2015 July 18-23; Washington DC, USA]. Alzheimers Dement 2015; 11 Suppl: P262-P263.
- 9. Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet 2015; 385: 2255-2263.
- 10. Anstey KJ, Eramudugolla R, Hosking DE, et al. Bridging the translation gap: from dementia risk assessment to advice on risk reduction. J Prev Alzheimers Dis 2015; 2: 189-198.
David Ames has received payments for consultancy from PranaBio, Pfizer, Janssen, Novartis, Eli Lilly and Lundbeck, and currently acts as principal site investigator for an investigational compound owned by Eli Lilly.