




Attention has now turned to combining new therapies to maximise their value
For decades, the 1-year overall survival for patients with advanced-stage or metastatic melanoma (stage IIIC unresectable or stage IV melanoma) was 30%–35%.1 It is now greater than 70%–80% in clinical trials of targeted and immune drug therapies (Box).2-10
This is good news for a cancer that continues to rise in incidence around the world and disproportionately affects the young. In Australia, melanoma is the fourth most common cancer and accounts for 10.1% of all new cases and 3.2% of cancer deaths.11 It is also the most common cancer in men aged 25 to 49 years, and women aged 15 to 24 years in New South Wales.12
Although incidence continues to rise in both sexes, mortality and 5-year survival remain stable,11 and the benefits observed with drug therapies in advanced-stage disease are yet to be reflected in population statistics. Most melanomas are cured with surgical excision: efforts are now focused on trialling novel active targeted and immune drug therapies in the 10%–15% of patients with early-stage melanoma that is predicted to recur and cause death.
Drug therapies
Melanoma is the poster child for immune and targeted approaches to cancer drug treatment. It is one of the few cancers where no chemotherapeutic agent has shown a survival benefit over best supportive care or single agent dacarbazine chemotherapy in patients with distant spread. Immunotherapies called checkpoint inhibitors and therapies targeting the mutated BRAF protein in the melanoma have put melanoma centrestage in the battle against cancer.
Immune therapy
Checkpoint inhibitors are a class of immune drugs that activate T cells for tumour-cell killing. Checkpoints are brakes on T cells to protect us from autoimmunity or an overzealous immune response to antigens, but can induce tolerance to cancer if activated. Ipilimumab is a fully human IgG1 monoclonal antibody that blocks the checkpoint called cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and was the first drug to show a survival advantage in advanced-stage melanoma compared with a vaccine or chemotherapy.7,8
The 1-year overall survival for patients treated with ipilimumab was 46% and 47% in each trial, respectively, although the response rates were only 11% and 15% (ie, the proportion of patients with at least a 30% reduction in tumour burden). Five-year survival in the latter study was 18.2% with ipilimumab, compared with 8.8% with chemotherapy.13
And yet, the relatively non-specific activation of the T cell induces autoimmune toxicities in 60% of patients. Although there have been reports of autoimmunity in almost every organ, the most frequent toxicities are dermatitis, inflammatory diarrhoea, endocrinopathies and hepatitis. It is critical that these autoimmune toxicities are identified and managed early to prevent complications.14
Biomarkers in the melanoma or host that enable us to select patients who are more likely to respond have been elusive, although recent research suggests specific neoantigens produced by the melanoma may be predictive.15 Predictive biomarkers would be particularly useful, not only to prevent toxicities in those who will not benefit, but also because responses may not be typical or may occur months after initiation of treatment.16
More recently, inhibitors of the programmed death 1 (PD1) checkpoint on T cells have shown high response rates (30%–40% in Phase I studies) and long survival durations with few autoimmune toxicities.9,17 Nivolumab and pembrolizumab are fully human or humanised IgG4 monoclonal antibodies, respectively, that inhibit the PD1 checkpoint. In the recently reported Phase III study of first-line nivolumab versus dacarbazine chemotherapy, the response rate was 40% and the 1-year overall survival was an impressive 73%.5 Autoimmune toxicities with both pembrolizumab and nivolumab occur in lower frequency and severity than with ipilimumab; most are easily managed, and resolve.
To increase the proportion of patients who benefit from immunotherapies, attention has turned to combining CTLA4 and PD1 checkpoint inhibitors, as each drug acts at a different time point during T-cell activation. In a Phase I study of such a combination, the response rate was over 50% at the recommended phase 3 dose, and the 1-year and 2-year survival rates across all dosing cohorts were estimated to be 85% and 79%, respectively;18 however, 53% of patients experienced at least one grade 3 or 4 autoimmune toxicity.3 Results of Phase III studies of ipilimumab versus nivolumab or pembrolizumab or ipilimumab plus nivolumab will be available in 2015. There are many other checkpoints on the T cell, and drugs targeting these additional points and other aspects of the immune system are in preclinical and clinical development.
Targeted therapy
In contrast, BRAF inhibitors are oral drugs that target the mutated BRAF protein in the melanoma cell. Mutated BRAF occurs in 40% of melanomas, and causes aberrant overactivation of a cell proliferation and survival pathway (the mitogen-activated protein kinase [MAPK] pathway). Treatment with BRAF inhibitors (dabrafenib or vemurafenib) results in rapid and deep shrinkage of metastases, including of brain metastases, in over 50% of patients, and tumour reduction of any amount occurs in over 85% of patients treated with these drugs.2,10
These drugs show a clear survival advantage over chemotherapy, with 1-year and 2-year overall survival rates of 70% and 46%, respectively.6 Adding a MEK inhibitor to a BRAF inhibitor backbone (MEK is the protein below BRAF in the MAPK pathway) results in further improvements in tumour shrinkage, progression-free survival4,19,20 and overall survival,21 and a reduction in the hyperproliferative cutaneous toxicities associated with BRAF inhibitor monotherapy.
How do we maximise response?
Primary resistance to immunotherapy is common; however, in those who respond, the response appears to be durable.9 In contrast, nearly all patients respond initially to targeted therapies, but more than 50% develop acquired resistance within 7 months of treatment with BRAF inhibitor monotherapy,22 and within 11 months of treatment with combined BRAF and MEK inhibitors.23 Compelling data show a brisk infiltration of clonal T cells into melanoma metastases early during treatment with MAPK targeted therapies,24 suggesting that this may be an opportune time to add immunotherapies to enhance the durability of response.
Translational research findings suggest that a cure may be possible for some patients with advanced-stage melanoma, and it may be realised by combining immune and targeted therapies. The pressing matters that now face us are how to sequence and combine these therapies to maximise patient outcomes, and how we recruit to clinical trials that seek to answer these questions as the single agents become available on the Australian Pharmaceutical Benefits Scheme for widespread use. Perhaps even more importantly, we may be able to prevent advanced-stage melanoma by using these therapies in early-stage disease, and trials are underway, although the question remains — how will the Australian community afford these drugs?25
Landmark overall survival rates for patients with advanced-stage melanoma (stage IIIC unresectable or stage IV melanoma) treated with immune or targeted therapies in Phase III clinical trials2-10
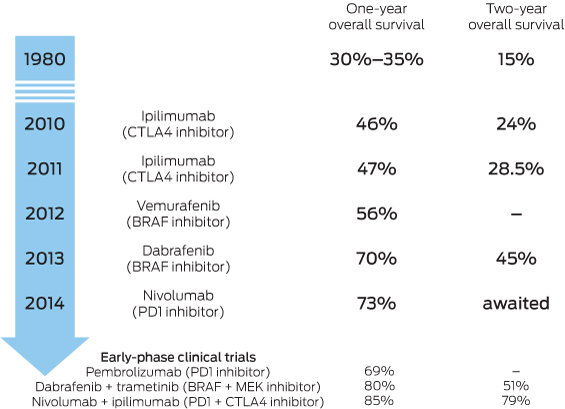
CTLA4 = cytotoxic T lymphocyte-associated antigen 4. BRAF/MEK = proteins in the mitogen-activated protein kinase pathway. When BRAF is mutated, it overactivates the pathway, causing uncontrolled cellular proliferation and survival. PD1 = programmed death 1 checkpoint.
Provenance: Commissioned; externally peer reviewed.
- 1. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009; 27: 6199-6206.
- 2. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380: 358-365.
- 3. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369: 122-133.
- 4. Flaherty K, Daud A, Weber JS, et al. Updated overall survival (OS) for BRF113220, a phase 1-2 study of dabrafenib (D) alone versus combined dabrafenib and trametinib (D+T) in pts with BRAF V600 mutation-positive (+) metastatic melanoma (MM) [abstract]. 2014 ASCO Annual Meeting. J Clin Oncol 2014; 32 (15 Suppl): 9010. http://meetinglibrary.asco.org/content/134903-144 (accessed Feb 2015).
- 5. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2014; 16 Nov [Epub ahead of print].
- 6. Hauschild A, Grobb J, Demidov LV, et al. An update on overall survival (OS) and follow-on therapies in BREAK-3, a Phase III, randomized trial: dabrafenib (D) vs. dacarbazine (DTIC) in patients (pts) with BRAF V600E mutation-positive metastatic melanoma (MM). [Poster presentation.] In: Abstract book of the 39th ESMO Congress (ESMO 2014) Madrid, Spain 26-30 September 2014. Ann Oncol 2014; 25 Suppl 4: iv374-iv393.
- 7. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711-723.
- 8. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364: 2517-2526.
- 9. Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32: 1020-1030.
- 10. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364: 2507-2516.
- 11. Australian Institute of Health and Welfare. Australia's health 2014. Canberra: AIHW, 2014. (AIHW Cat. No. AUS 178; Australia's Health Series No. 14.) http://www.aihw.gov.au/publication-detail/?id=60129547205&tab=2 (accessed Jan 2015).
- 12. Currow D, Thomson W. Cancer in NSW: incidence report 2009. Sydney: Cancer Institute NSW, Feb 2014. http://www.cancerinstitute.org.au/media/337679/cancer-in-nsw-incidence-report-2009.pdf (accessed Jan 2015).
- 13. Maio M, Bondarenko I, Robert C, et al. Survival analysis with 5 years of follow-up in a phase III study of ipilimumab and dacarbazine in metastatic melanoma [abstract]. European Cancer Congress; 2015 Sep 25–29; Vienna, Austria. http://2013.europeancancercongress.org/Scientific-Programme/Abstract-search.aspx?abstractid=7017 (accessed Feb 2015).
- 14. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 2012; 30: 2691-2697.
- 15. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371: 2189-2199.
- 16. Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15: 7412-7420.
- 17. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369: 134-144.
- 18. Wolchok JD, Kluger HM, Callahan MK, et al. Safety and clinical activity of nivolumab (anti-PD-1, BMS-936558, ONO-4538) in combination with ipilimumab in patients with advanced melanoma [abstract, and video of oral presentation (member access)]. 2013 ASCO Annual Meeting. J Clin Oncol 2013; 31 (15 Suppl): 9012. http://meetinglibrary.asco.org/content/107862-132 (accessed Feb 2015).
- 19. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014; 371: 1877-1888.
- 20. Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371: 1867-1876.
- 21. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015: 372: 30-39.
- 22. Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? Lancet Oncol 2014; 15: e371-e381.
- 23. Long GV, Fung C, Menzies AM, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun 2014; 5: 5694.
- 24. Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012; 18: 1386-1394.
- 25. Kefford RF. Drug treatment for melanoma: progress, but who pays? Med J Aust 2012; 197: 198-199. <MJA full text>
I acknowledge the funding support of the Sydney Medical School Foundation.
Georgina Long has consultant or advisory roles with Bristol-Myers Squibb, Roche, Merck Sharp & Dohme, GlaxoSmithKline, Novartis and Amgen. She has received honoraria from Bristol-Myers Squibb, Roche, Merck Sharp & Dohme and GlaxoSmithKline.