





Volume 201 - Issue 7
First Australian report of vitamin D-dependent rickets type I
Authors: Nobuaki Ito, Alexia S Peña, Shiree Perano, Gerald J Atkins, David M Findlay and Jennifer J Couper
Med J Aust 2014; 201 (7): 420-421. || doi: 10.5694/mja13.00220
Published online: 6 October 2014
Published online: 6 October 2014
Clinical findings and a homozygous mutation in the CYP27B1 gene demonstrate a rare form of vitamin D deficiency
A 20-month-old girl presented to hospital with features of rickets. Results of investigations were consistent with vitamin D-dependent rickets type I (VDDR-I), and DNA sequence analysis showed a homozygous mutation in the CYP27B1 gene of c.1325-1326insCCCACCC. This is the first reported Australian case of VDDR-I.
Clinical record
A 20-month-old white Australian girl presented to the Women's and Children's Hospital in Adelaide, South Australia, with a 6-month history of developmental regression of gross motor skills, failure to thrive and irritability, in particular distress when “she was wearing her shoes”. At 14 months of age, she had been crawling, pulling to stand and cruising, but at presentation her gross motor skills had regressed to only being able to sit unsupported. Other developmental milestones continued to progress normally for her age. Her weight at presentation was 7.7 kg, having dropped from the 50th centile to below the third centile (weight loss of 3.7 kg over 6 months). Length and head circumference had dropped from the 25th centile to less than the first centile and to the third centile, respectively, over this time.
The patient was the second child born to non-consanguineous white Australian parents. She was born at term with no perinatal complications. There was no history of familial hereditary disease. Maternal vitamin D deficiency had been treated during the pregnancy. The patient had received a multivitamin supplement (0.45 mL/day of Penta-vite [Bayer Australia], containing 10.1 µg of cholecalciferol) from birth until 2 months of age, with no further follow-up until her presentation at 20 months (Box 1).
On examination, the patient was mildly dehydrated, listless, miserable and mildly tachypnoeic, with intercostal recession. Her anterior fontanelle was large. Rachitic rosary, tibial bowing and widened metaphyses of the wrists and knees were evident. No bruising of the skin was noted.
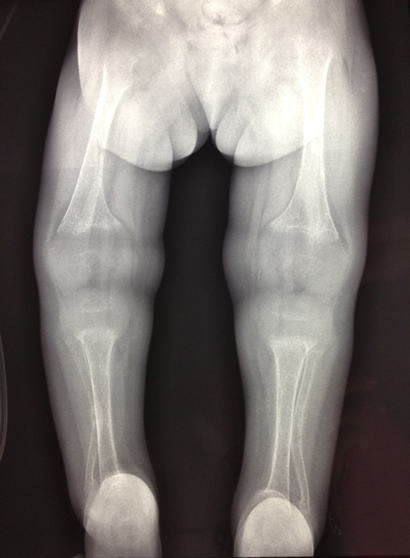
The patient had low serum total calcium and inorganic phosphate levels and a markedly elevated alkaline phosphatase level (Box 1). A skeletal survey showed significantly osteopenic bones, with flaring of the metaphyses in the long bones, soft tissue swelling around the elbow and wrist joints, and steep acetabular angles (Box 2). She had acute and healed fractures in multiple places: the right proximal ulnar shaft; both distal radial shafts; mid shafts of the second, third and fourth metacarpals of the right hand; mid shaft of the second metacarpal of the left hand; and mid shafts of the third metatarsals of both feet.
The clinical, radiological and biochemical findings were consistent with a diagnosis of rickets. She was treated with two doses of intravenous calcium infusion (12.5 mL/day of 10% calcium gluconate), oral phosphate (100 mg/kg/day) for 4 days, oral calcium (75 mg/kg/day) for 5 months, and ongoing calcitriol (0.07 µg/kg/day). She had feeding problems (food refusal and difficulty swallowing), which required nasogastric feedings for a month and a high-calorie diet.
The differential diagnosis included nutritional vitamin D-deficient rickets, vitamin D-dependent rickets (VDDR) type I and II, and hypophosphataemic rickets such as X-linked hypophosphataemia (Appendix 1). In the presence of normal renal function, electrolyte levels and acid–base balance, in addition to a low 1,25-dihydroxyvitamin D (1,25(OH)2D) level, normal 25-hydroxyvitamin D (25(OH)D) level, high alkaline phosphatase and parathyroid hormone (PTH) levels, and hypocalcaemia, the most likely diagnosis was VDDR type I (VDDR-I).
As VDDR-I is caused by a mutation in the CYP27B1 gene that impairs the conversion of 25(OH)D to 1,25(OH)2D, gene sequence analysis of the CYP27B1 gene was undertaken using genomic DNA from the index patient and her parents. All nine exons and intron–exon boundaries of CYP27B1 were amplified by polymerase chain reaction (PCR) from 100 ng of genomic DNA, using the PCR primer sets published previously.1 PCR conditions were 94°C for 5 min, followed by 35 cycles of amplification (94°C for 30 s, 54°C for 30 s, and 72°C for 30 s). The resulting PCR products were directly sequenced using Applied Biosystems 3730 and 3730xl DNA analysers. A biallelic c.1325-1326insCCCACCC sequence variation was found in exon 8 of the CYP27B1 gene (Appendix 2), which in turn led to a frameshift mutation and premature stop codon after 23 altered amino acids (F443PfsX466 or F443fs). As expected, a monoallelic c.1325-1326insCCCACCC sequence variation was found in both parents (Appendix 2). The parents have been counselled regarding their genetic status and the risk of having another child affected in an autosomal recessive manner.
Sixteen months after discharge, at 3 years of age, the patient demonstrated good catch-up in weight (12.6 kg; 10–25th centile), with a growth velocity of 7.8 cm/year (25–50th centile). She was walking unaided and walking up and down stairs. Metaphyseal widening and tibial bowing had decreased but were still present on clinical examination. She continued to receive calcitriol at a dose of 0.07 µg/kg/day, and her PTH, calcium and phosphate levels were within reference intervals (Box 1). A renal ultrasound produced normal results, with no evidence of nephrocalcinosis.
Discussion
We believe this is the first reported Australian case of VDDR-I with an identified mutation, demonstrated by a clinical presentation of severe rickets, severe hypophosphataemia, high PTH level, low 1,25(OH)2D level, failure to thrive and unresponsiveness to 25(OH)D therapy. The patient showed mild hypocalcaemia on admission due to the compensatory high PTH level. These findings are compatible with the current notion that the bone phenotype of rickets and osteomalacia mainly results from hypophosphataemia, not hypocalcaemia. Independent hypocalcaemia will not cause rickets or osteomalacia in hypoparathyroid patients, while hypophosphataemia alone causes rickets and osteomalacia in fibroblast growth factor 23 (FGF23)-related hypophosphataemia.2 The mutation of CYP27B1 in this case, c.1325-1326insCCCACCC, has been reported previously in diverse ethnic groups (Appendix 3).3-9 The final aberrant CYP27B1 protein lacks 44 amino acids in the C-terminus, resulting in reduced or no enzymatic activity to convert 25(OH)D into 1,25(OH)2D. This is the most common mutation among patients with VDDR-I; of 67 families with VDDR-I reported in the literature, including this one, 20 harbour this mutation. The index case is the first reported VDDR-I mutation in the Australian population, but ethnicity is irrelevant to the frequency or phenotype of the disease.
Previously described patients with VDDR-I have presented around the same age, with similar features (failure to thrive and regression in gross motor development) and similar results of biochemical investigations. Clinical data on the frequency of fractures in VDDR-I patients are limited due to the rarity of the disease.3-9 As there were no other causes found for the multiple fractures seen in our patient, these might have been related to the delay in diagnosis and treatment in a child who was increasing in mobility due to developmental progression. Almost all reported patients with VDDR-I have required regular calcitriol treatment. Although the required dose of calcitriol may depend on each patient, generally a dose of 0.01–0.1 µg/kg/day normalises serum calcium and inorganic phosphate levels in patients with VDDR-I.10
1 Results of laboratory investigations and calcitriol dose at each available time point
Patient's age | |||||||||||||||
Variable | RI | 1 day | 3 days | 2 months | 20 months (admission) | 21 months (discharge) | 25 months | 29 months | 32 months | 34 months | 37 months | ||||
Total calcium (mmol/L) | 2.10–2.65 | — | — | 2.36 | 2.00 | 2.32 | 2.20 | 2.33 | 2.31 | 2.32 | 2.21 | ||||
Inorganic phosphate (mmol/L) | 1.15–2.50 | — | — | 1.04 | 0.75 | 0.70 | 1.09 | 0.86 | 0.75 | 1.94 | 1.71 | ||||
Alkaline phosphatase (U/L) | 100–360 | — | — | 1078 | 3848 | 1494 | 969 | 1322 | 953 | 467 | 276 | ||||
Parathyroid hormone (pmol/L) | 0.5–5.5 | — | — | 32.9 | 37.0 | 33.1 | 15.2 | 29 | 14.8 | 3.7 | 2.3 | ||||
25(OH)D (nmol/L) | > 60 | 63 | 48 | 89 | 62 | — | — | — | — | — | — | ||||
1,25(OH)2D (pmol/L) | 50–160 | — | — | — | 17 | — | — | — | — | — | — | ||||
Calcitriol dose (µg/kg/day) | na | — | — | — | 0.01–0.04 | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | ||||
RI = reference interval. na = not applicable. — = data not available. 25(OH)D = 25-hydroxyvitamin D. 1,25(OH)2D = 1,25-dihydroxyvitamin D. | |||||||||||||||
Competing interests
No relevant disclosures.
Acknowledgements
This genetic work was supported by funding from the National Health and Medical Research Council Project Grants scheme.
References
- Alzahrani AS, Zou M, Baitei EY, et al. A novel G102E mutation of CYP27B1 in a large family with vitamin D-dependent rickets type 1. J Clin Endocrinol Metab 2010; 95: 4176-4183. 1
- Fukumoto S, Yamashita T. FGF23 is a hormone-regulating phosphate metabolism--unique biological characteristics of FGF23. Bone 2007; 40: 1190-1195. 2
- Yoshida T, Monkawa T, Tenenhouse HS, et al. Two novel 1alpha-hydroxylase mutations in French-Canadians with vitamin D dependency rickets type 1. Kidney Int 1998; 54: 1437-1443. 3
- Wang JT, Lin CJ, Burridge SM, et al. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am J Hum Genet 1998; 63: 1694-1702. 4
- Smith SJ, Rucka AK, Berry JL, et al. Novel mutations in the 1alpha-hydroxylase (P450c1) gene in three families with pseudovitamin D-deficiency rickets resulting in loss of functional enzyme activity in blood-derived macrophages. J Bone Miner Res 1999; 14: 730-739. 5
- Kitanaka S, Murayama A, Sakaki T, et al. No enzyme activity of 25-hydroxyvitamin D3 1alpha-hydroxylase gene product in pseudovitamin D deficiency rickets, including that with mild clinical manifestation. J Clin Endocrinol Metab 1999; 84: 4111-4117. 6
- Porcu L, Meloni A, Casula L, et al. A novel splicing defect (IVS6+1G>T) in a patient with pseudovitamin D deficiency rickets. J Endocrinol Invest 2002; 25: 557-560. 7
- Wang X, Zhang MY, Miller WL, et al. Novel gene mutations in patients with 1alpha- hydroxylase deficiency that confer partial enzyme activity in vitro. J Clin Endocrinol Metab 2002; 87: 2424-2430. 8
- Kim CJ, Kaplan LE, Perwad F, et al. Vitamin D 1alpha-hydroxylase gene mutations in patients with 1alpha-hydroxylase deficiency. J Clin Endocrinol Metab 2007; 92: 3177-3182. 9
- Durmaz E, Zou M, Al-Rijjal RA, et al. Clinical and genetic analysis of patients with vitamin D-dependent rickets type 1A. Clin Endocrinol (Oxf) 2012; 77: 363-369. lefthere