




Hepatitis C virus (HCV) infection progresses to chronic HCV in around 75% of cases,1 with the resultant risk of progressive liver fibrosis, cirrhosis and hepatocellular carcinoma. Around 20%–30% of people with chronic HCV will develop cirrhosis, generally following at least 20–30 years of infection.2 The large pool of chronic HCV in Australia (220 000)3 and the “ageing cohort” effect of this population related to high incidence of injecting drug use-acquired infection in the 1980s and 1990s means that the already escalating rates of HCV-related cirrhosis, liver failure and hepatocellular carcinoma are projected to further increase over the next two decades.4
Combined pegylated interferon (PEG-IFN) and ribavirin (RBV) treatment leads to a sustained virological response (SVR, equivalent to cure of infection) in around 50%–60% of people with chronic HCV, but treatment uptake remains low (3500–4000 people per year).5 Several factors contribute to low HCV treatment rates, including toxicity of interferon-based therapy, prolonged course of treatment (24–48 weeks), social marginalisation of many people with chronic HCV infection, lack of treatment infrastructure (particularly in opiate pharmacotherapy, prison, community health, and primary care settings), and lack of awareness of the curative potential of treatment. Poorer HCV treatment responses in those with advanced liver disease also limit the impact on disease burden.
Fortunately, a revolution in HCV treatment is fast approaching, with the advent of direct-acting antiviral (DAA) therapy and the move towards interferon-free regimens.6 Before the end of this decade, simple (single daily dosing oral regimens), tolerable, short-duration (6–24 weeks) therapy with extremely high efficacy (cure rates above 90%) should be the norm for the HCV-infected population. The broad implementation of such therapeutic regimens has the potential to produce one of the major turnarounds in disease burden seen in public health and clinical medicine.
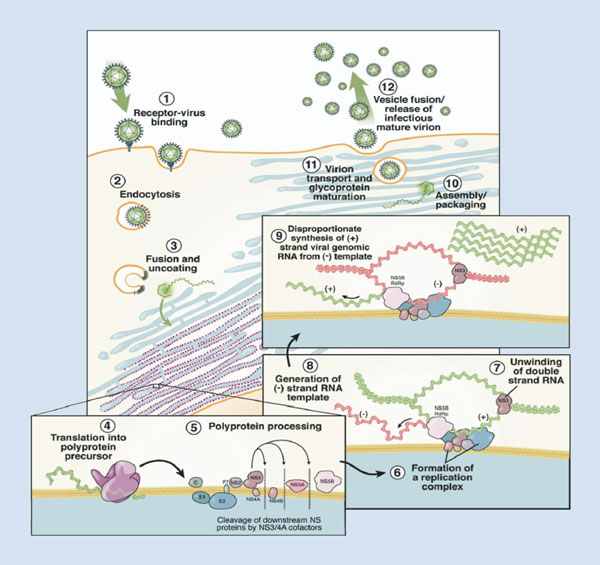
As the HCV viral life cycle has been more fully elucidated (Box 1),7 rational drug design has been used to identify small molecule inhibitors of various HCV proteins involved in HCV replication: (i) NS3/4A protease, which is involved in post-translation processing of HCV polyproteins; (ii) NS5B RNA-dependant RNA polymerase that is required for copying the HCV RNA genome and transcribing mRNA; and (iii) NS5A enzyme, which like the two previous target enzymes is involved in HCV viral replication, although its functions are somewhat less clear.
Employing drug development strategies similar to those used for HIV antiretroviral therapy, many inhibitors of these three target enzymes are in Phase 2 and 3 clinical development, and two HCV protease inhibitors have been licensed (Box 2).
Over the past 12 months, several major milestones have been reached in the clinical development of DAA therapy for chronic HCV infection. Findings from Phase 3 trials in treatment-naive and treatment-experienced patient populations with genotype 1 (55% of the chronic HCV population in Australia8) have been published for two HCV protease inhibitors, telaprevir and boceprevir.9-13 In treatment-naive populations, telaprevir or boceprevir, when added to PEG-IFN/RBV (triple therapy), improve SVR (undetectable HCV RNA 24 weeks post-treatment) from 38%–44% to 66%–75%, and enable treatment duration to be shortened from 48 weeks to 24–28 weeks for around half of patients.9,11 In treatment-experienced populations, telaprevir and boceprevir both provided considerably enhanced SVR when combined with PEG-IFN/RBV (from 17%–21% to 64%–66%), although treatment response varied from around 30% for those patients with a prior “null response” (< 2 log decline in HCV RNA with PEG-IFN/RBV through Week 12), to around 55% for those with a prior “partial response” (> 2 log decline HCV RNA at Week 12, but no on-treatment undetectable HCV RNA), to 75%–85% for those with prior relapse (end-of-treatment undetectable HCV RNA with post-treatment viral rebound).10,12 Telaprevir and boceprevir were licensed by the United States Food and Drug Administration in May 2011. However, several concerns regarding these two agents remain, including:
a problematic dosing schedule of three times per day with a meal (fatty for telaprevir), and a large pill burden (boceprevir, 12 pills per day; telaprevir, six pills per day);
reduced treatment efficacy in patients with prior PEG-IFN/RBV null response, particularly those with advanced fibrosis (10%–20% SVR);
high rates of HCV resistance in patients with limited IFN sensitivity — for many patients with prior PEG-IFN/RBV null response, the addition of a single protease inhibitor provides functional monotherapy;
therapeutic toxicity, including increased incidence of rash (telaprevir) and anaemia (boceprevir and telaprevir) — anaemia is more pronounced with boceprevir, due to the longer duration of protease inhibitor therapy (24–44 weeks, compared with 12 weeks for telaprevir);
a large number of potential drug–drug interactions, due to the metabolism of HCV protease inhibitors through cytochrome P450 (3A4) pathways;
complex treatment algorithms, with different strategies for telaprevir (12 weeks of triple therapy followed by 12–36 weeks of PEG-IFN/RBV treatment) and boceprevir (a 4-week PEG-IFN/RBV “lead in”, followed by 24–44 weeks of triple therapy), and different strategies for patients with cirrhosis and non-cirrhosis;
complex HCV RNA monitoring schedules and stopping rules that differ between telaprevir and boceprevir; and
the high cost of protease inhibitor agents — in the US, boceprevir is $1100 per week ($26 400–$48 400 for a treatment course) and telaprevir is $49 200 for a 12-week course — which will be added to cost of PEG-IFN/RBV therapy.
Telaprevir and boceprevir also have more limited potency against non-1 HCV genotypes, with particularly poor activity against genotype 3,14,15 and have therefore only been licensed for genotype 1.
Assuming these two initial HCV protease inhibitors receive PBAC listing, appropriate selection of patients for PEG-IFN/RBV/telaprevir and PEG-IFN/RBV/boceprevir regimens will be crucial. In the HCV-treatment-naive population, many of the pretreatment factors that are predictive of PEG-IFN/RBV response remain predictive for response to protease inhibitor-containing triple therapy, in particular fibrosis stage.9,11 Host genetic polymorphisms in the IL28B region, recently associated with PEG-IFN/RBV response in chronic HCV genotype 1 infection,16-18 also remain predictive of triple therapy response19 albeit less so due to the overall higher response rates.
Factors that are specific to DAA therapy response should also be helpful in patient selection. HCV genotypes 1a and 1b are not associated with PEG-IFN/RBV treatment response, but in protease inhibitor-containing triple therapy, patients with genotype 1a have a higher rate of treatment failure, which is related to the lower genetic barrier to viral resistance compared with genotype 1b.20
Triple therapy with PEG-IFN/RBV/protease inhibitor (telaprevir or boceprevir) should be the standard of care for patients with chronic HCV genotype 1 infection for the next 2–3 years. However, the rapid clinical development of numerous DAA agents across several classes provides the opportunity for both improved triple therapy regimens and additional therapeutic strategies. HCV protease inhibitors with preliminary evidence of enhanced efficacy, minimal toxicity, and improved dosing schedules (daily dosing)21,22 are in Phase 3 evaluation in combination with PEG-IFN/RBV (TMC435, BI 201335).
Quadruple therapy, with PEG-IFN/RBV and two DAA agents, is being evaluated in patients with prior PEG-IFN/RBV null and partial response and in those with advanced fibrosis. A small Phase 2 study in genotype 1 patients with prior PEG-IFN/RBV null response using PEG-IFN/RBV combined with BMS-650032 (protease inhibitor) and BMS-790052 (NS5A inhibitor) for 24 weeks produced a 100% (all 10 patients) SVR rate.23 This compares extremely favourably with the SVR of around 30% seen in prior PEG-IFN/RBV null responders treated with triple therapy as described above.
IFN-free HCV therapy was initially evaluated in the INFORM-1 study undertaken in Australia and New Zealand, which demonstrated that mericitabine/danoprevir (nucleoside analogue/protease inhibitor) combination produced potent viral suppression with no viral breakthrough during 14 days of dual dosing.24 Following this proof of concept, a study explored the curative potential of IFN-free dual DAA therapy. In patients with chronic HCV genotype 1 and prior PEG-IFN/RBV null response, four of 11 treated with BMS-650032 (protease inhibitor) and BMS-790052 (NS5A inhibitor) for 24 weeks achieved an SVR.23 Of particular interest, both genotype 1b patients were cured compared with two of nine genotype 1a patients. A subsequent study involving Japanese chronic HCV genotype 1b-only patients treated with the same regimen provided a 100% SVR rate (all 10 patients).25
Important data have also been presented on IFN-free therapy for chronic HCV genotype 2 and 3a. A Phase 2 study undertaken in New Zealand evaluated the nucleotide analogue PSI-7977 in combination with RBV for 12 weeks, with a 100% SVR rate (all 10 patients).26 The high genetic barrier to resistance, cross-genotype activity, and preliminary evidence of limited toxicity, make PSI-7977 (now known as GS-7977) a very attractive agent for use with other DAA agents and/or RBV. Further patient cohorts are evaluating the GS-7977/RBV regimen for chronic HCV genotype 1. Phase 3 data will be required to confirm preliminary efficacy and, more importantly, to evaluate safety in larger study populations.
Several other interferon-free combination DAA regimens are under clinical evaluation.27 A combination of a nucleos(t)ide analogue with an NS5A inhibitor may be an ideal therapeutic strategy as both classes have cross-genotype activity and lack the CYP3A4 interactions seen with the HCV protease inhibitor class.
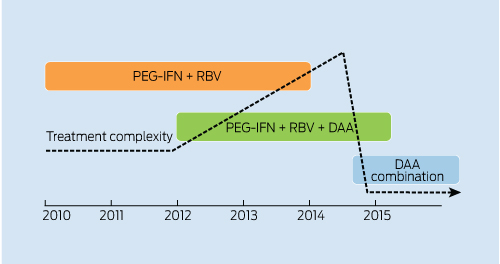
Although time lines for future HCV therapeutic strategies are not completely clear, initial Phase 3 studies of IFN-free therapy will commence in 2012. Thus, it is possible that the period of triple therapy with PEG-IFN/RBV and protease inhibitor therapy will be relatively short-lived, to be replaced by IFN-free therapy for the vast majority of patients with HCV (Box 3). Initial concerns with telaprevir- and boceprevir-containing triple therapy, as outlined above, will increase the overall complexity of HCV clinical management. However, the likely switch to IFN-free regimens with markedly reduced toxicity, shortened treatment durations, and simplified dosing and monitoring schedules should lead to a rapid decline in complexity (Box 3). Clearly, significant toxicity or suboptimal efficacy revealed in Phase 3 evaluation with various IFN-free regimens will delay this relatively optimistic time line. Broad availability within the Australian context will also be dependent on the PBAC approval process.
The initial phase of DAA-based therapy will provide enhanced efficacy, but challenges to their clinical utility will be evident. There are several priorities for the next 3–5 years as we move towards IFN-free simplified combination DAA regimens. Chronic HCV should continue to be treated primarily as a chronic liver disease with enhanced capacity for staging of fibrosis. Non-invasive methods of liver disease staging, in particular hepatic elastography (Fibroscan), will be central to this strategy. The recent development of portable Fibroscan technology should further improve access to disease staging in non-tertiary clinic settings. Those patients with at least moderate fibrosis (estimated 54 000)3 should be targeted for therapeutic intervention, given their significant risk of progression to advanced liver disease complications. Once the era of IFN-free combination DAA therapy arrives, the overall strategy should switch to treatment of HCV as predominantly an infectious disease involving therapeutic intervention for all stages of disease (including acute HCV). Patients with advanced liver disease will continue to require specific liver disease management, while the utility of IFN-free therapy in the setting of HCV-related liver failure (including potential hepatic function restorative capacity) remains unknown. A major increase in HCV treatment uptake will require expanded access through community-based and primary care practices, opiate pharmacotherapy clinics, and prisons. Furthermore, enhanced public HCV awareness and targeted screening campaigns will be required. The potential for HCV treatment as a prevention tool, a remote possibility in the IFN-based therapy era, may also become a feasible public health strategy.
Provenance: Commissioned; externally peer reviewed.
- Gregory J Dore1
- The Kirby Institute for infection and immunity in society, University of New South Wales, Sydney, NSW.
I am a member of the Roche, Bristol-Myers Squibb, Merck Sharp and Dohme, Gilead, and Janssen advisory boards, and have received payment for travel scholarships, speakers’ bureaus and research grants from these companies.
- 1. Micallef JM, Kaldor JM, Dore GJ. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J Viral Hepat 2006; 13: 34-41.
- 2. Thein HH, Yi Q, Dore GJ, Krahn MD. Estimation of stage-specific fibrosis progression rates in chronic hepatitis C virus infection: a meta-analysis and meta-regression. Hepatology 2008; 48: 418-431.
- 3. The Kirby Institute. HIV, viral hepatitis and sexually transmissible infections in Australia Annual Surveillance Report 2011. Sydney: The Kirby Institute, the University of New South Wales, 2011. http://www.med.unsw.edu.au/NCHECRweb.nsf/resources/2011/$file/KIRBY_ASR2011.pdf (accessed Mar 2012).
- 4. Razali K, Thein HH, Bell J, et al. Modelling the hepatitis C virus epidemic in Australia. Drug Alcohol Depend 2007; 91: 228-235.
- 5. Gidding HF, Topp L, Middleton M, et al. The epidemiology of hepatitis C in Australia: notifications, treatment uptake and liver transplantations, 1997-2006. J Gastroenterol Hepatol 2009; 24: 1648-1654.
- 6. Asselah T, Marcellin P. New direct-acting antivirals’ combination for the treatment of chronic hepatitis C. Liver Int 2011; 31 Suppl 1: 68-77.
- 7. Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132: 1979-1998.
- 8. Dore GJ, Law M, MacDonald M, Kaldor JM. Epidemiology of hepatitis C virus infection in Australia. J Clin Virol 2003; 26: 171-184.
- 9. Poordad F, McCone J Jr, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364: 1195-1206.
- 10. Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364: 1207-1217.
- 11. Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364: 2405-2416.
- 12. Zeuzem S, Andreone P, Pol S, et al; REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364: 2417-2428.
- 13. Sherman KE, Flamm SL, Afdhal NH, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365: 1014-1024.
- 14. Imhof I, Simmonds P. Genotype differences in susceptibility and resistance development of hepatitis C virus to protease inhibitors telaprevir (VX-950) and danoprevir (ITMN-191). Hepatology 2011; 53: 1090-1099.
- 15. Foster GR, Hézode C, Bronowicki JP, et al. Telaprevir alone or with peginterferon and ribavirin reduces HCV RNA in patients with chronic genotype 2 but not 3 infections. Gastroenterology 2011; 141: 881-889.
- 16. Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41: 1100-1104.
- 17. Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461: 399-401.
- 18. Rallón NI, Soriano V, Naggie S, et al. IL28B gene polymorphisms and viral kinetics in HIV/hepatitis C virus-coinfected patients treated with pegylated interferon and ribavirin. AIDS 2011; 25: 1025-1033.
- 19. Chayama K, Hayes CN, Abe H, et al. IL28B but not ITPA polymorphism is predictive of response to pegylated interferon, ribavirin, and telaprevir triple therapy in patients with genotype 1 hepatitis C. J Infect Dis 2011; 204: 84-93.
- 20. Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53: 1742-1751.
- 21. Dieterich D, Asselah T, Guyader D, et al. SILEN-C3: Treatment for 12 or 24 weeks with BI201335 combined with peginterferon alfa-2a and ribavirin (P/R) in treatment-naive patients with chronic genotype-1 HCV infection. Hepatology 2011; 54 Suppl: 378A.
- 22. Fried M, Buti M, Dore GJ, et al. TMC435 in combination with peginterferon and ribavirin in treatment-naive HCV genotype 1 patients: final analysis of the PILLAR phase IIb study. Hepatology 2011; 54 Suppl: 1429A.
- 23. Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366: 216-224.
- 24. Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376: 1467-1475.
- 25. Chayama K, Takahashi S, Kawakami Y, et al. Dual oral combination therapy with the NS5A inhibitor BMS-790052 and the NS3 protease inhibitor BMS-650032 achieved 90% sustained virological response (SVR12) in HCV genotype 1b-infected null responders. Hepatology 2011; 54 Suppl: 1428A.
- 26. Gane E, Stedman CA, Hyland RH, et al. Once daily PSI-7977 plus RBV: pegylated interferon-alfa not required for complete rapid viral response in treatment-naive patients with HCV GT2 or GT3. Hepatology 2011; 54 Suppl: 377A.
- 27. Gane E. Future hepatitis C virus treatment: interferon-sparing combinations. Liver Int 2011; 31 Suppl 1: 62-67.
Summary
The next decade will be a crucial period in the public health response to hepatitis C virus (HCV) infection.
The rapid development of direct-acting antiviral therapy for HCV infection has brought considerable optimism to the HCV sector, with the realistic hope that therapeutic intervention will soon be more effective and offer shorter treatment duration.
The initial phase of combination pegylated interferon, ribavirin and a protease inhibitor will be associated with increased toxicity and complexity of therapeutic management but, over the course of the decade, strategies including interferon-free combination direct-acting antiviral regimens with enhanced tolerability and simplified dosing schedules and monitoring protocols will emerge.