





Volume 195 - Issue 6
Blurred vision and pain in the eye
Authors: Michael H Barnett, Gurjit Chohan and Leo Davies
Med J Aust 2011; 195 (6): 329-332. || doi: 10.5694/mja11.10707
Published online: 19 September 2011
Published online: 19 September 2011
Summary
Subacute unilateral visual impairment accompanied by pain on eye movement is characteristic of optic neuritis.
Most cases of optic neuritis resolve spontaneously, and acute treatment with intravenous steroids hastens recovery but does not alter the ultimate visual outcome.
Brain magnetic resonance imaging (MRI) may permit a diagnosis of multiple sclerosis (MS) to be made after a single clinical demyelinating event such as optic neuritis.
Current evidence supports the introduction of disease-modifying therapy in patients with a single clinical event such as optic neuritis and brain MRI compatible with MS.
The diagnosis of MS is a confronting life event associated with significant personal, social and financial burdens. The diagnosing neurologist should provide a detailed explanation of the disease and its clinical spectrum and introduce the patient to the wide range of support services, educational material and MS clinics.
Grading system for recommendations in Practical neurology*
Grade A: “Excellent” — evidence from large randomised controlled trials (RCTs), or systematic reviews with meta-analyses of multiple RCTs
Grade B: “Good” — one or more RCTs with limitations, or high-quality and persuasive non-randomised cohort studies
Grade C: “Satisfactory or poor” — non-randomised case series and opinions of experts
* Adapted from the National Health and Medical Research Council.
Mary’s story
Mary, who is 28 years old, presented to her general practitioner after developing a dull headache and eye pain over 3 days. The pain was localised to her right periorbital region and forehead, and was made worse by eye movement. Soon after onset of the headache, she noticed blurred vision in her right eye; in particular, coloured objects appeared pale or washed out compared with their appearance through her left eye. This worsened progressively over the 3 days — vision through her right eye became markedly reduced, grey and blurred. She had no significant prior medical history.
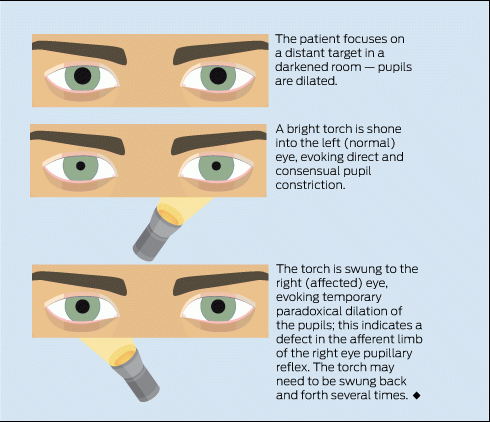
On examination, Mary’s right eye and orbit appeared normal, but her visual acuity was 6/6 in the left eye and 6/12 in the right eye. Her pupils were equal in size. Direct and consensual pupillary responses were reduced when light was directed at her right eye. The right pupil appeared to paradoxically dilate when the torch was swung alternately from the left to the right eye. On ophthalmoscopy, the cornea, anterior chamber, lens and vitreous humour appeared normal in both eyes, as did both optic discs. There were no haemorrhages or other abnormal retinal findings. Mary’s eye movements were normal, but she found looking to the extremes of gaze painful in the right eye. No other abnormal neurological findings were detected.
Approach to the problem
Interpreting the clinical findings
Subacute history: The evolution of symptoms over days in this case strongly suggests an inflammatory process. In contrast, a vascular event in the optic nerve would be expected to result in acute symptoms, and a neoplastic process usually evolves more slowly.
Pain on eye movement: The presence of pain on eye movement is non-specific but suggests an inflammatory process affecting the globe, optic nerve or orbital contents.
Diminution of colour intensity: Pale, washed-out vision with relatively little loss of visual acuity is characteristic of processes involving the optic nerve and chiasm.
Unilateral visual impairment: Visual impairment in one eye only localises the abnormality to either the eye or the optic nerve. The “swinging flashlight test” detects a difference in the visual stimulus from each eye that reaches the efferent limb of the pupillary reflex when the two eyes are stimulated alternately (Box 1). In this test, paradoxical dilation of the pupil on direct light stimulation indicates a relative afferent pupillary defect (Marcus Gunn pupil) and is pathognomonic of a process that affects the optic nerve, or less commonly the retina, in one eye. Conversely, this test is usually unaffected by conditions that interfere with the transmission of light through the eye itself, such as corneal injury, cataract and processes that make the anterior chamber or the vitreous humour cloudy.
Absence of optic disc swelling: While a unilateral swollen optic nerve head definitively localises the abnormality to that optic nerve, its absence, which suggests that the pathological process is retrobulbar, is common in inflammatory optic neuropathy. Bilateral swelling of the optic nerve heads most commonly reflects raised intracranial pressure, or may be a rare manifestation of bilateral simultaneous anterior inflammatory optic neuropathy.
Differential diagnoses
Differential diagnoses of unilateral visual impairment localised clinically to the optic nerve are summarised in Box 2.
Optic neuritis, an immune-mediated inflammation of the optic nerve, is the most likely cause of Mary’s constellation of symptoms and signs. It most commonly occurs in the context of multiple sclerosis (MS) or as an idiopathic isolated event. The other cause of inflammatory optic neuritis, neuromyelitis optica (NMO), is rare and far less likely. Current NMO diagnostic criteria require both optic neuritis and myelitis, although the presence of specific serum autoantibodies may raise suspicion of NMO at the time of initial presentation with an index event such as optic neuritis.
While a compressive lesion should always be considered in patients with optic neuropathy, the short temporal evolution of the symptoms argue against this possibility. Other causes of optic neuropathy — such as ischaemia, diabetes, sarcoidosis and rare infectious diseases — are unlikely in a young woman with no relevant medical history and, in comparison to idiopathic optic neuritis, are more likely to affect the anterior portion of the nerve and produce visible swelling on ophthalmoscopy.
Appropriate use of investigations
Optic neuritis is primarily a clinical diagnosis, and investigations are largely adjunctive. Clinical features that would make a diagnosis of optic neuritis unlikely are listed in Box 3.
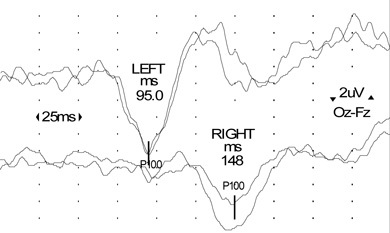
Visual evoked potentials (VEPs): In a typical clinical presentation of optic neuritis, as in this case, recording VEPs is not mandatory, but the presence of a delayed potential with preserved morphology helps confirm the diagnosis of demyelination (Box 4).
Blood tests: Blood tests are extremely low-yield investigations in a patient with typical symptoms and signs of optic neuritis, but full blood count, tests for erythrocyte sedimentation rate (ESR), fasting glucose level and angiotensin-converting enzyme level, and screening for serum autoantibodies are routinely performed to help exclude rare causes of optic neuropathy and inflammatory diseases of the central nervous system (CNS). If a patient has atypical, bilateral or recurrent optic neuritis, NMO may be a possible diagnosis and a serum NMO-IgG assay (available in many teaching hospital laboratories in Australia) should be performed to test for the presence of specific anti-aquaporin 4 antibodies.
Magnetic resonance imaging (MRI): Brain MRI is the most helpful investigation, as it provides aetiological and prognostic information. In patients with a “clinically isolated syndrome” (a single clinical demyelinating event such as optic neuritis) and MRI findings consistent with more widespread demyelination, the risk of a recurrent symptomatic demyelinating episode over the subsequent decade — and hence clinically definite MS — is approximately 80%.1 The diagnosis of MS requires the dissemination of lesions in both space and time. Although a second clinical attack remains the “gold standard” for dissemination in time, new diagnostic criteria (the 2010 revisions to the McDonald criteria,2 which incorporate information obtained from MRI to show dissemination in time and place) permit a diagnosis of MS to be made after a single clinical demyelinating event.
Analysis of cerebrospinal fluid: Cerebrospinal fluid examination for oligoclonal bands (which are present in 90%–95% of patients with MS, but are not specific for the condition) is not necessary in a patient with clinical and MRI features that are typical of MS. It may be useful in patients with atypical clinical or MRI features.
Mary was referred urgently to a neurologist, with a view to urgent MRI. VEPs showed significant prolongation of the cortical potential (P100) latency, indicating slowed conduction in the right optic nerve consistent with a demyelinating process. Results of a full blood count, tests for ESR, fasting glucose level and angiotensin-converting enzyme level, and screening of serum for autoantibodies were unremarkable.
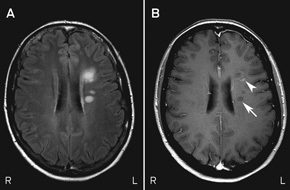
MRI scans of Mary’s brain and orbits showed altered signal intensity within the retrobulbar portion of the right optic nerve. They also showed six small areas of altered signal intensity in the supratentorial white matter affecting the periventricular and subcortical regions, one of which exhibited enhancement with the contrast agent gadolinium (Box 5). The simultaneous presence of enhancing and non-enhancing brain lesions (ie, actively inflamed and older lesions) is sufficient information to prove dissemination in time, and Mary could be told that she has MS without waiting for a second attack.
Therefore, based on the 2010 McDonald criteria for MS, the final diagnosis for Mary was optic neuritis due to MS (Box 6).
Acute therapy
Optic neuritis usually resolves spontaneously over weeks to months, and simple analgesia and observation are often sufficient during the acute phase. In severe attacks (visual acuity 6/60 or worse), or when a patient has an occupational or other need to recover vision faster than the natural history of the condition, consideration can be given to the use of high-dose intravenous methylprednisolone (usually administered on an outpatient basis as 1 g/day over 3–5 days), which rapidly reduces pain, limits inflammation and hastens functional recovery (Grade A evidence).4
However, there is no difference in the ultimate visual outcome in patients treated with intravenous steroids versus observation alone,5 thus use of such therapy is controversial. Results of the pivotal Optic Neuritis Treatment Trial suggest that administration of moderate-dose oral prednisone (60 mg/day) to patients with acute optic neuritis increases the risk of recurrence and should therefore be avoided (Grade A evidence).4
Disease-modifying therapy
The initiation of disease-modifying therapy in patients with a clinically isolated episode of optic neuritis and a diagnosis of MS based on MRI is controversial. There is an increasing body of clinical, neuropathological and MRI evidence to suggest that there may be a window of opportunity during which immunomodulatory therapies, such as interferon β (subcutaneous or intramuscular) and glatiramer acetate (subcutaneous), should be commenced. In part, this has been confirmed by randomised controlled trials in which the initiation of such therapy has been shown to reduce the risk of developing clinically definite MS at 2–3 years by up to 50% (Grade A evidence),6 and reduce disability at 3 years (Grade B evidence),7 compared with delayed treatment. While long-term data favouring the treatment of patients with a clinically isolated syndrome are lacking, there is mounting circumstantial evidence that links inflammation in early MS with chronic axonal loss in the later stages of the disease, supporting the early introduction of immunomodulatory therapy.
Fact or fiction?
FACT: It is true that, in patients with a single clinical demyelinating event (such as optic neuritis) and magnetic resonance imaging findings that are consistent with more widespread demyelination, the risk of a recurrent symptomatic demyelinating episode over the subsequent decade — and hence clinically definite multiple sclerosis — is approximately 80%.
FICTION: It is not true that intravenous steroid therapy in patients with acute optic neuritis improves long-term visual outcomes and prevents the development of optic atrophy.
Disease-modifying treatments are expensive, and prescribing these for patients with a clinically isolated syndrome (such as a single episode of optic neuritis) in Australia is currently not subsidised by the Pharmaceutical Benefits Scheme (PBS). Despite the Therapeutic Goods Administration granting approval for this indication, and the capacity to diagnose many patients with MS at their first presentation, the PBS currently only subsidises treatment for “clinically definite MS”, which, by definition, requires two clinical attacks.
The therapeutic options for managing MS have been expanded by the advent of new disease-modifying agents including natalizumab, which is administered as a monthly intravenous infusion, and fingolimod, the first oral agent for the condition.8,9 While considered more efficacious than conventional immunomodulatory treatment, these agents have short- and medium-term risks associated with immunosuppression and systemic effects. Natalizumab therapy can, rarely, be associated with progressive multifocal leukoencephalopathy (PML), a potentially fatal viral infection of the CNS. The risk of PML is determined partly by treatment duration, previous exposure to immunosuppressive therapies and the presence of serum antibodies to the causative agent, JC virus. Fingolimod, a novel immunosuppressive agent, has rare short-term cardiovascular and ophthalmic side effects (bradyarrhythmia and macular oedema, respectively); the risk of potential long-term hazards, such as serious opportunistic infection, is unknown.
At present, the risk–benefit analysis for disease-modifying therapy in patients with a single clinical demyelinating event, such as optic neuritis, weighs in favour of initiating conventional therapy with interferon β or glatiramer acetate. Disease-modifying therapy should be maintained throughout the relapsing phase of MS (which has a highly variable duration that, without therapy, averages 10 years), and escalation to fingolimod or natalizumab should be considered in patients who continue to exhibit clinical disease activity despite conventional treatment. Periodic review (6–12-monthly) by a neurologist is advisable to determine whether escalation of therapy is appropriate. Subclinical disease activity may also merit escalation of therapy and can be monitored for with periodic brain MRI, but the utility of routine neuroimaging in asymptomatic patients is not well established.
For women with MS, early pregnancy (the first two trimesters) confers moderate protection against relapse, and stable MS should not be considered a disincentive to starting a family. Although there are no data suggesting an adverse effect of conventional immunomodulatory therapy on the developing human fetus, it is recommended that these agents be withdrawn before conception. Pregnancy should therefore ideally be planned in concert with the GP, the treating neurologist and, where relevant, the MS nurse.
Support for the patient
The diagnosis of MS carries a significant personal, social and financial burden. Although newly diagnosed patients are often young and internet-savvy, the diagnosing neurologist is best placed to quell anxiety with a detailed explanation of the disease and its clinical spectrum. Data on the natural history of MS suggest that 50% of patients will require assistance to walk within 15 years of diagnosis, but 10%–15% of patients never accrue significant disability. Patients whose initial presentation is with optic neuritis may have a milder disease course. It is likely that the early introduction of disease-modifying therapy will favourably alter long-term outcomes. Substantial support services are available, including MS Australia programs for newly diagnosed patients (http://www.mssociety.org.au/just-diagnosed.asp) and those receiving immunotherapy (http://www.mssociety.org.au/immunotherapy-support.asp), and a range of educational materials are available from MS Australia and from pharmaceutical companies. The integration of MS nurse specialists into MS clinics in Australia has transformed the management of patients with newly diagnosed disease. MS nurse specialists are the primary point for patient contact; they provide counselling, coordinate the implementation of services and deliver ongoing immunotherapy support.
Mary’s visual acuity had spontaneously recovered to 6/6 in the affected eye at follow-up 6 weeks after presentation. Minor impairment of right eye colour perception persisted. She subsequently commenced subcutaneous interferon β-1b, 250 μg every 2 days, prescribed by her neurologist and initiated in her home by MS Australia nursing staff. She was advised to use adequate contraception while taking interferon β-1b. Her injection technique was reviewed in the MS clinic 8 weeks later, at which stage she had mild interferon-related flu-like symptoms that were ameliorated by paracetamol. At 6 months, there had been no further discrete neurological attacks, results of a neurological examination were normal and brain MRI showed no new or enhancing (active) lesions. Mary continued to work full time as a sales consultant and had no immediate plans to become pregnant. Six-monthly reviews in the MS clinic were scheduled, with instructions for Mary to contact the MS nurse specialist if she developed any new, sustained neurological symptoms in the interim.
2 Differential diagnoses of unilateral visual impairment localised clinically to the optic nerve
Optic neuritis (immune-mediated inflammation of the optic nerve) can occur in isolation or can be related to multiple sclerosis
Compressive optic neuropathy can be secondary to a tumour (benign or malignant) or secondary to a carotid-ophthalmic aneurysm
Ischaemic optic neuropathy may be arteritic (usually in older patients, and there may be other features of giant cell arteritis) or non-arteritic (more common in patients with hypertension, diabetes mellitus, a history of smoking, and small optic discs with a small cup-to-disc ratio)
Malignant infiltration of the optic nerve (by glioma, leukaemia, lymphoma or carcinoma) merits consideration in cases that are atypical (eg, visual impairment that evolves over weeks to months)
Optic neuropathy of other causes including diabetes (diabetic papillopathy), neuromyelitis optica (NMO or Devic’s disease, a rare inflammatory condition that commonly results in severe visual loss), granulomatous optic neuropathy due to sarcoidosis, systemic autoimmune diseases (eg, Sjögren’s syndrome or systemic lupus erythematosus), and rare infectious diseases (eg, cat-scratch disease, Lyme disease, toxoplasmosis or syphilis)
3 Atypical features of unilateral visual impairment that suggest a diagnosis other than idiopathic optic neuritis
Gradually progressive visual impairment over weeks to months*
Absence of pain on eye movement
Age > 50 years and/or known vascular risks
Severe optic disc swelling*
Bilateral optic disc swelling*
Preserved colour vision
Absence of a relative afferent pupillary defect
Extensive vitreous cellular reaction on slit lamp examination*
4 Visual evoked potentials
5 Magnetic resonance imaging scans of Mary’s brain
6 Pathophysiology of multiple sclerosis (MS)
MS is a multifocal inflammatory demyelinating disorder of the central nervous system (CNS) with an unknown aetiology. Both genetic and environmental factors contribute to MS susceptibility and underlie the epidemiological variability of the disease. In Australia, MS has a prevalence of about 70 per 100 000 population. The disease is most common in Tasmania and least common in northern Queensland. This “latitudinal gradient” may be explained by putative, but unproven, environmental factors such as sunlight exposure, vitamin D levels and Epstein–Barr virus infection. Genetic susceptibility is conferred primarily by the presence of a specific human leukocyte antigen allele (HLA-DRB1*1501 in people of European ancestry), but more than 50 other gene loci that confer a smaller risk for MS have been identified. MS has an average age of onset in the early 30s and, like many diseases that involve the immune system, is more common in women. The most widely accepted view of its aetiology is an autoimmune attack directed against normal constituents of the CNS (“outside-in” hypothesis), but there is also evidence of a primary pathological process affecting glial cells, with secondary recruitment of an adaptive immune response (“inside-out” hypothesis). Actively inflamed lesions are characterised by destruction of myelin, oligodendrocytes and, to a lesser extent, axons.
In relapsing MS, the clinical features correlate with the anatomic localisation of lesions, such as the optic nerve. Most cerebral lesions are clinically silent, but those occurring in “eloquent areas”, such as the optic nerve and spinal cord, are usually symptomatic. The symptoms that accompany optic neuritis and other MS lesions are caused by focal inflammation, impaired axonal conduction due to demyelination, and axonal loss. Recovery, which is often incomplete, is the rule in early relapsing MS and is largely attributable to resolution of inflammation and remyelination. However, there is no discernible regeneration of axons in the CNS, and loss of axons correlates with the development of irreversible disability.3
With increasing disease duration, areas of new focal inflammation become less common and remyelination progressively fails. Global changes affecting the white matter, characterised by activation of microglia and nerve fibre loss, become prominent. This shift in the pathophysiology over the course of the disease has implications for the timing of anti-inflammatory therapy.
Competing interests
Michael Barnett has served on scientific advisory boards for Merck Serono, Novartis, and Sanofi-Aventis, and has received research support from Multiple Sclerosis Research Australia and the Nerve Research Foundation (University of Sydney); his institution has received unencumbered research grants from Bayer, Biogen Idec, Novartis and Sanofi-Aventis. Gurjit Chohan is a recipient of a Merck Serono Multiple Sclerosis Fellowship at the Brain and Mind Research Institute.
References
- Brex PA, Ciccarelli O, O’Riordan JI, et al. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med 2002; 346: 158-164. 0_CIGGCBEE
- Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69: 292-302. 0_HHGHHEEE
- Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338: 278-285. 0_i1140317
- Beck RW, Cleary PA, Anderson MM Jr, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med 1992; 326: 581-588. 0_i1140319
- Beck RW, Cleary PA, Backlund JC. The course of visual recovery after optic neuritis. Experience of the Optic Neuritis Treatment Trial. Ophthalmology 1994; 101: 1771-1778. 0_i1140321
- Coyle PK. Early treatment of multiple sclerosis to prevent neurologic damage. Neurology 2008; 71: S3-S7. 0_i1140323
- Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet 2007; 370: 389-397. 0_i1140325
- Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010; 362: 387-401. 0_i1140327
- Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2003; 348: 15-23. 0_i1140330
Shifting focus to adolescent wellbeing and inclusive participation in the digital age
Allyson R Todd, Elena Wang, Stephanie R Partridge
The clinical presentation, investigation, and management of women diagnosed with endometriosis in Australian general practices, 2011–2021: an open cohort study
Danielle Mazza, Kailash Thapaliya, Sharinne B Crawford, Alissia Hui, Maryam Moradi, Luke E Grzeskowiak
The impact of the Breast Screen NSW transition from film to digital mammography, 2002–2016: a linked population health data analysis
Rachel Farber, Nehmat Houssami, Katy J L Bell
Non‐technical errors associated with deaths in surgical care, Australia, 2012–2019, by surgical specialty (Australian and New Zealand Audit of Surgical Mortality): a retrospective cohort study
Jesse Ey, Victoria Kollias, Octavia Lee, Kelly Hou, Matheesha Herath, John B North, Ellie Treloar, Suzanne Edwards, Martin Bruening, Adam J Wells, Guy J Maddern
The CURE Asthma roadmap
Gary P Anderson, Anthony Flynn, Phil G Bardin, John D Blakey, Shyamali C Dharmage, Paul Foster, Peter G Gibson, Adam Jaffe, Alan James, Christine R Jenkins, Sundram Sivamalai, Peter D Sly, Guy B Marks, Vanessa M McDonald, Judy Wetttenhall
Evidence from Australian cohort studies about asthma trajectories and transitions across the life course: a narrative review
Caroline Lodge, Xin Dai, Ingrid A. Laing, Michael P Menden, Anthony Flynn, Gary P Anderson, Sarath Ranganathan, Shyamali C Dharmage