





Volume 195 - Issue 3
Why is treating obesity so difficult? Justification for the role of bariatric surgery
Author: Joseph Proietto
Med J Aust 2011; 195 (3): 144-146. || doi: 10.5694/j.1326-5377.2011.tb03242.x
Published online: 1 August 2011
Published online: 1 August 2011
Abstract
There is little evidence that public health measures adopted so far have had any impact on the rise in the prevalence of obesity.
Weight-loss programs have a very high long-term failure rate.
There is emerging evidence that weight is regulated by the hypothalamus and is physiologically defended.
There is also a strong genetic predisposition to the development of obesity.
The availability and promotion of high-energy foods and the absence of any obligatory need for physical activity compound the problem, but this social change is not easily reversible.
One way forward is to focus public health measures on preventing obesity in children while making resources available to treat people who are already obese, including providing funding for bariatric surgery in public hospitals.
The prevalence of obesity is rising in many countries.1,2 On the surface, it would seem entirely reasonable to adopt public health measures to combat a disorder that is widespread and of high prevalence. This approach has succeeded in controlling epidemics of infectious diseases and in reducing the incidence of smoking. Why then is obesity proving so much more difficult to control? This question cannot be answered without first understanding the regulation of body weight and the aetiology of obesity.
The regulation of body weight
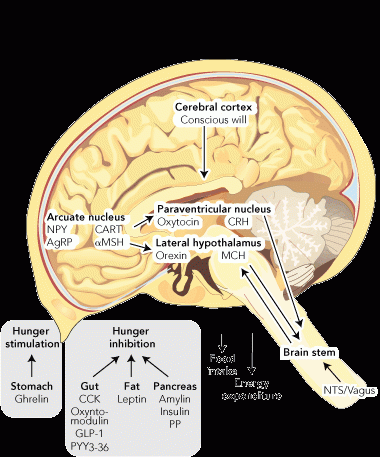
Weight is regulated by the hypothalamus (Box 1). In the arcuate nucleus there are neurons that produce neuropeptide Y and agouti-related peptide, both of which powerfully induce hunger. In the same area, other neurons produce cocaine and amphetamine-regulated transcript (CART) and pro-opiomelanocortin (which is cleaved to form melanocyte-stimulating hormone [MSH]). Both CART and MSH suppress the desire to eat. Thus whether someone feels the need to eat or not depends on the balance of the activity of these “first order” neurons. They, in turn, project to other areas of the hypothalamus that also receive input from the brain stem via the nucleus of the tractus solitarius, which transmits signals received from the vagus nerve.
This central mechanism is modulated by circulating hormones, which can influence the desire to eat. These come from the gastrointestinal tract, the pancreas, and adipose tissue. They can be divided into stimulators and inhibitors of hunger. So far, only one stimulator of hunger has been discovered — ghrelin, which is made in the stomach. In contrast, there are many inhibitors of hunger, including: cholecystokinin, oxyntomodulin, glucagon-like peptide-1 and peptide YY, all from the intestine; insulin, amylin and pancreatic polypeptide from the pancreas; and leptin from adipose tissue.
Causes of obesity
The recent increase in the prevalence of overweight and obesity is blamed on the continuous availability of high-energy foods, together with a major reduction in the obligatory need for physical activity. However, not everyone becomes obese when placed in an obesogenic environment. Genetically lean individuals will become overweight, but not obese, when placed in this environment. Obesity is prevented by an increase in leptin levels as fat accumulates. This is a classical negative feedback system. For the environment to produce obesity, there needs to be a genetic susceptibility that impairs this negative feedback system. Hence, when young people are overfed by the same amount, some put on weight while others do not.3 Results from twin and adoption studies have shown a genetic predisposition to weight gain.4-7 Overfeeding monozygotic twins results in a range of weight gain, however, the amount of weight gained within each twin pair is very closely correlated.8 While our genes may be responsible for a significant proportion of obesity (defined by a body mass index > 30 kg/m2),4 it is clear that monogenetic mutations that lead to severe obesity, such as leptin deficiency9 or melanocortin-4 receptor mutations,10 are rare and cannot explain most cases of obesity. In addition, the recent increase in the prevalence of obesity cannot be the result of change at a genetic level, because mutations accrue over a much longer time frame. The emerging evidence that much of obesity may be epigenetic in origin may reconcile these two apparently opposing views.
Epigenetic obesity
Epigenetic modification is the permanent change in gene expression caused by environmental factors that induce chemical modifications, such as methylation of the promoter and/or alterations in the way DNA is packaged.11,12 There has been a longstanding belief that epigenetic modifications occur mainly in utero. However, there is some emerging evidence that early postnatal nutritional status can also influence epigenetic gene regulation.13 Susceptible rats made obese by feeding, an energy-rich diet for 12 weeks after weaning defend their elevated weight after diet-induced weight loss, even when they are subsequently exposed only to low-fat rat chow ad libitum.14
The physiological defense of body weight
After weight loss, changes in both energy expenditure and in hunger-controlling hormones encourage weight regain. For example, after weight loss, ghrelin levels rise15 while leptin, cholecystokinin and insulin levels fall.16 In addition, the thyroid hormone T4 is converted to the inactive reverse-T3 instead of T3,17 contributing to the reduction in energy expenditure. It is likely that it is these physiological adaptations that make it so difficult to maintain weight loss. Importantly, if this regulatory mechanism is operating in those who are already obese, public health messages encouraging people to eat healthy food and to exercise are unlikely to have long-term impact on their weight.
The high failure rate of obesity management
Several studies have shown that, although many obese people who make the effort can achieve and maintain significant weight loss for 1–2 years, the weight is usually regained over the longer term. Box 2 lists some of the studies that followed patients for more than 3 years, all showing that weight loss was largely not maintained.
In contrast, Box 3 shows the long-term outcomes of weight loss as a result of bariatric surgery, ranging from 13% to 31% even after 10 years of observation.21-24
Bariatric surgery should be made available as part of a comprehensive weight loss service in public hospitals. The model adopted by the Weight Control Clinic at Austin Health is that patients undergo an intensive medical program first, and are referred for surgery only if that approach fails. Weight loss reverses or ameliorates many of the comorbid conditions associated with obesity, including type 2 diabetes, metabolic syndrome, obstructive sleep apnoea, and infertility. Several studies have shown that bariatric surgery in obese patients with type 2 diabetes is cost-effective. Indeed, an Australian study has concluded that bariatric surgery produces both cost savings and health benefits.25
Conclusion
What is the solution to improving the management of obesity? First, we must no longer ignore the scientific evidence. It appears that once someone becomes obese, that state is physiologically defended. This newly discovered biology explains the high failure rate of obesity management. If secondary prevention is difficult as a result, we must focus our attention on primary prevention and stop children from becoming obese. Finally, we must help the long-suffering obese in their struggle to maintain a reduced weight. In the absence of safe, effective pharmacological agents that can be used long-term, bariatric surgery is the most successful intervention for sustained weight loss. Why is it not more often conducted in public hospitals?
1 Weight regulation and influences on the desire to eat
 | |||||||||||||||
|
AgRP = agouti-related peptide. CART = cocaine and amphetamine-regulated transcript. CCK = cholecystokinin. CRH = corticotropin-releasing hormone. GLP-1 = glucagon-like peptide. MCH = melanin-concentrating hormone. αMSH = alpha melanocyte-stimulating hormone. NPY = neuropeptide Y. NTS = nucleus of the tractus solitarius. PP = pancreatic polypeptide. PYY = peptide YY. | |||||||||||||||
2 Studies of dietary approaches to long-term weight loss in overweight or obese individuals who were followed for at least 3 years
Study |
Design |
||||||||||||||
Wadden et al18 |
Studied 76 women who were randomly allocated to a very-low energy diet (VLED) alone, behaviour modification alone, or both. The combined therapy group lost the most weight (16.8 kg; 15.8%). However, 5 years after treatment, most people in all three groups had returned to their pretreatment weight. |
||||||||||||||
Walsh et al19 |
Studied 145 overweight patients who were treated with a VLED. Initial weight losses were 27.2 kg (22.0%) for men and 19.3 kg (18.8%) for women. At 54 months after entering the program, the average maintained loss was 5.1 kg (5.5%). |
||||||||||||||
Anderson et al20 |
A meta-analysis of 29 studies that aimed to examine the long-term weight-loss maintenance of individuals completing a structured weight-loss program. It concluded that at 4–5 years, the average weight loss maintenance was 7.1 kg (6.6%) for VLED studies and 2.0 kg (2.1%) for diet studies. |
||||||||||||||
3 Studies of bariatric surgery for weight loss in overweight or obese individuals who were followed for 3–10 years
Study |
Number of patients |
Bariatric procedure |
Total weight lost |
Duration of observation |
Weight loss at end of observation period |
||||||||||
Sjöström et al21 |
34 |
Roux-en-Y gastric bypass |
38% |
10 years |
25% |
||||||||||
Sjöström et al21 |
156 |
Gastric banding |
21% |
10 years |
13% |
||||||||||
Sugerman et al22 |
1025 |
Roux-en-Y gastric bypass |
35% |
10 years |
28% |
||||||||||
Waters et al23 |
157 |
Roux-en-Y gastric bypass |
36% |
3 years |
30% |
||||||||||
Yale24 |
251 |
Roux-en-Y gastric bypass |
36% |
5 years |
31% |
||||||||||
Competing interests
I was chair of the Optifast medical advisory committee for Nestle until 2010. I was a member of the expert panel that developed guidelines for bariatric surgery for the treatment of type 2 diabetes.
Acknowledgements
Many thanks to Priya Sumithran and Scott Baker who read and offered helpful advice on the manuscript. I am supported by the Sir Edward Dunlop Medical Research Foundation.
References
- Wang Y, Beydoun MA, Liang L, et al. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity 2008; 16: 2323-2330. i1096010
- Cameron AJ, Welborn TA, Zimmet PZ, et al. Overweight and obesity in Australia: the 1999–2000 Australian Diabetes, Obesity and Lifestyle Study (AusDiab). Med J Aust 2003; 178: 427-432. i1096012
- Levine JA, Eberhardt NL, Jensen MD. Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science 1999; 283: 212-214. i1096014
- Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA 1986; 256: 51-54. i1096016
- Stunkard AJ, Sorensen TI, Hanis C, et al. An adoption study of human obesity. N Engl J Med 1986; 314: 193-198.
- Stunkard AJ, Harris JR, Pedersen NL, McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med 1990; 322: 1483-1487.
- Sorensen TI, Holst C, Stunkard AJ, Skovgaard LT. Correlations of body mass index of adult adoptees and their biological and adoptive relatives. Int J Obes Relat Metab Disord 1992; 16: 227-236. i1096020
- Bouchard C, Tremblay A. Genetic effects in human energy expenditure components. Int J Obes 1990; 14 Suppl 1: 49-58. i1096022
- Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997; 387: 903-908. i1096024
- Stutzmann F, Tan K, Vatin V, et al. Prevalence of melanocortin-4 receptor deficiency in Europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes 2008; 57: 2511-2518. i1096026
- Gluckman PD, Hanson MA. Developmental and epigenetic pathways to obesity: an evolutionary-developmental perspective. Int J Obes (Lond) 2008; 32 Suppl 7: S62-S71. i1096028
- van Vliet J, Oates NA, Whitelaw E. Epigenetic mechanisms in the context of complex diseases. Cell Mol Life Sci 2007; 64: 1531-1538. i1096030
- Waterland RA, Lin JR, Smith CA, Jirtle RL. Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum Mol Genet 2006; 15: 705-716. i1096032
- Levin BE, Dunn-Meynell AA. Defense of body weight against chronic caloric restriction in obesity-prone and -resistant rats. Am J Physiol Regul Integr Comp Physiol 2000; 278: R231-R237. i1096034
- Cummings DE, Weigle DS, Frayo RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 2002; 346: 1623-1630. i1096036
- Chearskul S, Delbridge E, Shulkes A, et al. Effect of weight loss and ketosis on postprandial cholecystokinin and free fatty acid concentrations. Am J Clin Nutr 2008; 87: 1238-1246. i1096038
- Vagenakis AG, Portnay GI, O’Brian JT, et al. Effect of starvation on the production and metabolism of thyroxine and triiodothyronine in euthyroid obese patients. J Clin Endocrinol Metab 1977; 45: 1305-1309. i1096040
- Wadden TA, Sternberg JA, Letizia KA, et al. Treatment of obesity by very low calorie diet, behavior therapy, and their combination: a five-year perspective. Int J Obes 1989; 13 Suppl 2: 39-46. i1096042
- Walsh MF, Flynn TJ. A 54-month evaluation of a popular very low calorie diet program. J Fam Pract 1995; 41: 231-236. i1096044
- Anderson JW, Konz EC, Frederich RC, Wood CL. Long-term weight-loss maintenance: a meta-analysis of US studies. Am J Clin Nutr 2001; 74: 579-584. i1096046
- Sjöström L, Lindroos AK, Peltonen M, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351: 2683-2693. i1096048
- Sugerman HJ, Wolfe LG, Sica DA, Clore JN. Diabetes and hypertension in severe obesity and effects of gastric bypass-induced weight loss. Ann Surg 2003; 237: 751-756. i1096050
- Waters GS, Pories WJ, Swanson MS, et al. Long-term studies of mental health after the Greenville gastric bypass operation for morbid obesity. Am J Surg 1991; 161: 154-157. i1096052
- Yale CE. Gastric surgery for morbid obesity. Complications and long-term weight control. Arch Surg 1989; 124: 941-946. i1096054
- Keating CL, Dixon JB, Moodie ML, et al. Cost-effectiveness of surgically-induced weight loss for the management of type 2 diabetes: modeled lifetime analysis. Diabetes Care 2009; 32: 567-574. i1096058
Provenance: Not commissioned; externally peer reviewed.