





Volume 195 - Issue 10
Practical Neurology Part 5: Recurrent unresponsive episodes and seizures
Authors: Melissa A DeGruyter and Mark J Cook
Med J Aust 2011; 195 (10): 586-591. || doi: 10.5694/mja11.11260
Published online: 21 November 2011
Published online: 21 November 2011
Janice’s story: Janice, who is 23 years old, presented to her general practitioner with a history of stereotyped episodes that her partner had observed every 1-2 weeks over the previous year. She described the episodes as starting with “butterflies in the stomach”(rising up to her throat and lasting about 20 seconds) and intense deja vu.
Summary
Careful history-taking is essential when evaluating patients with suspected epileptic seizures. It should focus on ascertaining whether the episodes are seizures or a seizure mimic such as syncope.
Recurrent unresponsive episodes associated with seizures may indicate a diagnosis of focal epilepsy or complex partial epilepsy.
Adults with a clinical diagnosis of a focal seizure disorder require investigation with electroencephalo-graphy and magnetic resonance imaging.
The goal of treatment should be to achieve a life free of seizures, with minimum adverse effects from anticonvulsant medication. The choice of medication should be individualised to a patient’s seizure characteristics, circumstances and preferences. Dose adjustments should be made according to clinical response (seizure frequency and adverse effects), rather than on serum drug concentrations alone.
Lifestyle advice, such as advice about driving restrictions, is important for the safety of the patient and others.
All anticonvulsants are potentially teratogenic. Poorly controlled epilepsy in pregnancy imparts significant risks to the mother and baby, which need to be weighed against the risks of teratogenicity. The risk of major congenital malformations is highest with valproate, particularly in high doses.
Janice’s story
Janice, who is 23 years old, presented to her general practitioner with a history of stereotyped episodes that her partner had observed every 1–2 weeks over the previous year. She described the episodes as starting with “butterflies in the stomach”(rising up to her throat and lasting about 20 seconds) and intense deja vu. Her partner reported that Janice then suddenly becomes unresponsive, has a blank look, and makes chewing movements with her mouth. Occasionally, she would pick at her clothes with her left hand and hold her right arm in an odd fixed posture. The episodes would typically last 1–2 minutes, but it would be several more minutes before Janice became fully responsive and she had no memory of the episodes beyond the premonitory sensations. On two occasions, the episodes progressed to a witnessed generalised tonic–clonic seizure of 1–2 minutes that terminated spontaneously. She was confused for several minutes after both these episodes, one of which involved turning of her head to the right.
Janice’s medical history was unremarkable apart from a prolonged febrile seizure at 11 months of age, and results of a neurological examination were nomal. Her only medication was the oral contraceptive pill. She worked in a clerical position.
Approach to the problem
Interpretation of history and examination
Although the history of episodic altered awareness with abnormal movements and behaviour suggests a diagnosis of focal epilepsy, other conditions must be excluded (Box 1). The first step is to consider whether the episodes result from convulsive electrical activity in the cerebral cortex, as opposed to other causes of acute behavioural change.
Careful history-taking, including a detailed account of the circumstances and behaviour before, during and after the episode, is essential. As patients almost invariably have limited recall of events during a seizure, a witness account is essential. Since epilepsy carries substantial psychosocial impact, the diagnosis should not be made unless the clinician has a high degree of certainty.
Aura: Rising epigastric sensations and deja vu commonly occur in auras of seizures that have a focal onset. An aura is a “simple” partial seizure — a manifestation of epileptic activity confined to a small portion of the brain, without impairment of awareness or memory. Once a seizure includes brain structures that are involved in awareness and memory, it is described as “complex”. Auras can occur in isolation or can precede complex partial seizures. Auras are not a feature of primarily generalised seizures — patients typically have no warning of the onset of these, which rapidly engage both hemispheres of the brain.1 Auras may provide localising information: seizures arising from the temporal lobe result in deja vu, fear or a rising epigastric sensation; somatosensory auras, manifesting as paraesthesia, may be reported by patients with parietal lobe epilepsy; and visual auras, such as static, flashing, or moving lights or images, are characteristic of seizures arising from the occipital lobe.
Behaviour: A detailed description of behaviour during an episode helps with diagnosis. During complex partial seizures (also known as “focal” or “localisation-related” seizures), patients typically stop what they are doing (behavioural arrest) and stare motionlessly for 30 seconds to a few minutes. Another common feature of complex partial seizures is automatism — repetitive purposeless movement, commonly involving the mouth (eg, chewing motions, lip smacking) or one or both hands (eg, fidgeting, picking at clothes). Focal seizures originate in a discrete area of the brain but tend to spread rapidly over a few seconds to affect adjacent cortex.1 Thus a simple partial seizure can progress to a complex partial seizure and then a generalised convulsive seizure — a process known as secondary generalisation.2
Characteristics of suspected seizures: In Janice’s case, the abnormal behaviour preceding the convulsions implies a focal event that became secondarily generalised. Some of the lateralising features, namely dystonic limb posturing and versive head movements (ie, forced conjugate ocular and cephalic and/or truncal deviation), typically occur contralateral to the seizure focus. Similarly, new neurological signs such as aphasia or hemiparesis, seen transiently in the postictal state, provide powerful localising information. Postictal confusion and drowsiness, lateral tongue bite, urinary incontinence, myalgia (typically the next day) and headache are strong indicators of a seizure, rather than a seizure mimic such as syncope.3 However, convulsive motor activity and urinary incontinence can occur in both seizures and syncope, so a diagnosis of syncope should not be excluded based on these features alone.
Neurological examination: Results of a neurological examination are often normal in people with epilepsy, as in Janice’s case, but occasionally there are findings that may point to a diagnosis: examples include limb asymmetry due to perinatal stroke or the stigmata of phacomatoses such as tuberous sclerosis. Recent-onset, lateralising, neurological abnormalities indicate the possibility of a structural brain lesion such as a cerebral neoplasm or an arteriovenous vascular malformation.
Appropriate use of investigations
Once epileptic seizures are identified, an investigative plan is required. In Janice’s case, the clear history of complex partial events means that a focal lesion, such as a neoplasm or scar, should be ruled out. As her symptoms were longstanding (ie, a year or more), an aggressive lesion (eg, tumour) is unlikely, but such a diagnosis should be considered in patients with a brief history of seizures or new “fixed” neurological signs.
Electroencephalography: An electroencephalogram (EEG) should generally be performed when evaluating a patient with an unprovoked seizure.4 While the results of interictal EEGs are often normal, even when the diagnosis of a seizure disorder has been established, an EEG may provide information regarding localisation and seizure type. Normal EEG results do not rule out a seizure disorder, and some people have epileptiform abnormalities on EEG but never go on to have a clinical seizure disorder. The frequency of abnormalities on EEG may be increased by sleep deprivation or provocative strategies such as hyperventilation, although the diagnostic yield of such measures is uncertain. Video EEG monitoring, where the EEG recording is accompanied by continuous video recording of the patient, may help characterise seizures and explore the possibility of a diagnosis other than a seizure disorder in patients who have frequent episodes.
Neuroimaging: Nearly all adult patients with newly diagnosed focal seizures should undergo neuroimaging to assess for structural lesions of the brain, such as cerebral tumours, arteriovenous malformations, or scarring of the temporal lobe (hippocampal sclerosis). Magnetic resonance imaging (MRI) is the modality of choice, as it is superior to computed tomography in demonstrating subtle tissue changes such as those associated with an old ischaemic insult.4 However, it is essential that any structural findings are interpreted with caution and correlated with the clinical seizure characteristics and EEG findings, as structural lesions may be incidental (eg, diffuse white matter disease, lacunar infarcts, calcification, vascular spaces). Functional imaging modalities (such as positron emission tomography and single-photon emission computed tomography) can be used to evaluate patients who have medically refractory epilepsy and are being considered for surgical treatment, as these may provide additional localising information. Imaging may not be appropriate in children with typical benign focal syndromes, such as benign rolandic epilepsy and occipital epilepsy.
Blood tests: There is no clear evidence for the routine use of blood tests for determining a cause of seizures other than epilepsy (eg, hyponatraemia, uraemia, hepatic encephalopathy, cerebral infection). Blood tests may be useful in patients whose clinical presentations suggest a specific underlying problem. Minor degrees of hypomagnesaemia, hypocalcaemia and hypoglycaemia may be detected but are rarely relevant in such cases.
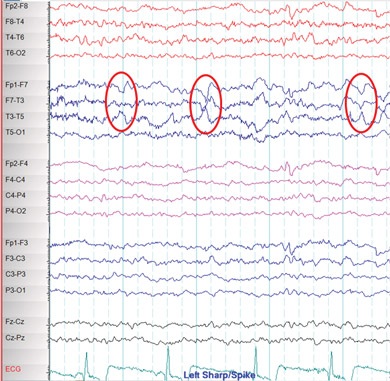
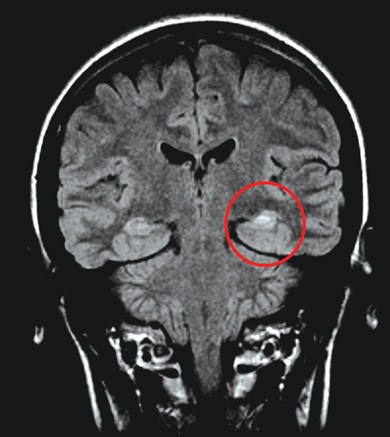
Janice was referred to a neurologist, who ordered an EEG and high-resolution MRI of her brain. The interictal EEG showed spikes of electrical activity in the left temporal lobe (Box 2) with predominance in the anterior temporal region. The MRI revealed atrophy of the left hippocampus with an associated increase in signal intensity (Box 3).
Based on these findings and the clinical history, Janice was diagnosed with complex partial epilepsy and hippocampal sclerosis affecting the mesial left temporal lobe.
Hippocampal sclerosis is the most commonly identified pathological lesion in mesial temporal lobe epilepsy. A history of febrile seizures in infancy may be a marker of the condition in a minority of patients, such as Janice. Whether hippocampal sclerosis is a cause or an effect of seizures is uncertain.2
Management
The key goal of managing epilepsy is optimal quality of life with no seizures and minimal adverse effects from medications. The consequences of uncontrolled epilepsy can be severe and include injury, social disability and increased risk of death.
Identifying the epilepsy syndrome correctly has implications for therapy and investigations; the idiopathic generalised epilepsies have a distinctive EEG pattern, do not require structural imaging (as they are not related to structural abnormalities) and may be aggravated by some anticonvulsants, particularly carbamazepine. They differ too in prognosis — most idiopathic epilepsies of childhood go into spontaneous remission by the late teenage years.
Fact or fiction?
Fact: It is true that the doses of anticonvulsants should be titrated according to clinical response, including seizure frequency and signs of drug toxicity, rather than serum drug concentrations.
Fiction: It is not true that a normal electroencephalogram (EEG) result excludes a diagnosis of epilepsy — most patients with epileptic seizures will have a normal interictal EEG result, and a normal EEG result does not rule out a seizure disorder.
Fiction: It is not true that high-dose preparations of the oral contraceptive are reliably effective when a patient is also taking hepatic enzyme-inducing anticonvulsant medication.
Anticonvulsant therapy
In the past 20 years, a variety of new drugs for treating epilepsy have become available, expanding choices but complicating decisions about which anticonvulsant to use (Box 4). In addition to seizure type and epilepsy syndrome, one needs to consider the availability, cost, adverse effects and potential interactions of the various anticonvulsants, as well as the patient’s age, sex, comorbidities, other medications and preference, in choosing the type and dose of anticonvulsant.
Systematic reviews comparing the efficacy of older drugs (carbamazepine, phenytoin, valproate, phenobarbitone and primidone) in treating partial seizures have found no significant differences between drugs (Grade A evidence),5 with the exception of a meta-analysis that identified a small advantage of carbamazepine over valproate (Grade A evidence).6 While none of the newer drugs (oxcarbazepine, gabapentin, lamotrigine, topiramate, levetiracetam, tiagabine, pregabalin and lacosamide) have been shown to have efficacy that is superior to carbamazepine, phenytoin or valproate, better tolerability has been shown for some (Grade B evidence).7 Current evidence-based clinical guidelines recommend carbamazepine or phenytoin as first-line agents for adults with focal epilepsy, as there is no clear first choice medication based on efficacy data alone (Grade B evidence).5
The potential for bone disease to be caused by anticonvulsants — by both agents that induce hepatic enzymes (eg, phenytoin, carbamazepine) and those that do not (eg, valproate) — has been shown (Grade B evidence).8 The effects of the newer agents on bone have not been established (Grade C evidence).8 Monitoring the bone health of patients on anticonvulsants is a controversial issue, but patients who are at high risk of bone disease (eg, older patients, patients with osteoporosis, and those with a strong family history of osteoporosis) may need monitoring with regular bone densitometry and early introduction of therapy to prevent bone loss (Grade B evidence).8
Anticonvulsant therapy normally begins with a single agent at a low dose. The dose is gradually increased until seizures are controlled or adverse effects arise. Dose adjustments are made according to clinical response, including seizure frequency and signs of toxicity (eg, sedation, ataxia, diplopia), rather than serum drug concentrations alone. If the first agent is ineffective or poorly tolerated, a second is added and the first is then tapered and discontinued over several weeks. While monotherapy is the preferred goal, combination therapy may be required in patients who do not have adequate seizure control after trying two or three different agents. In such cases, referral to an epilepsy specialist or epilepsy centre is appropriate.
Patients who do not gain sufficient control of their seizures with anticonvulsant medication should be referred to a neurologist with a special interest in epilepsy. The nature of the episodes and the syndromic diagnosis should be confirmed, and other therapies should be considered (eg, surgery, other invasive interventions).
As Janice was experiencing disabling and distressing seizures, she was commenced on carbamazepine. Potential adverse effects (eg, rash, dizziness, diplopia, blurred vision, sedation) were explained and she was informed that she could not drive until she had attained a seizure-free period of 6 months. Carbamazepine was introduced slowly to minimise adverse effects — 100 mg twice daily for 2 weeks, then 200 mg twice daily. Janice was advised to avoid bathing and swimming unless supervised because of safety concerns should a seizure occur.
Lifestyle advice
It is important to establish whether a patient has an occupation (eg, driver of a public vehicle or plane) or hobby (eg, activities involving height or water) that provides particular risk and to individualise advice on this. In Australia, patients with newly diagnosed epilepsy can return to driving after a seizure-free period of 6 months from the start of therapy, or 3 months on the recommendation of an experienced consultant. This applies to private drivers only; guidelines for commercial licenses are more complex.9 These national regulations are currently under review. The Austroads website (http://www.austroads.com.au) provides more detail on licensing requirements.
When Janice was reviewed 3 months later, she reported better control of her seizures in terms of frequency and no secondary generalised events, but she was still experiencing monthly complex partial seizures and frequent auras. Although free of any adverse effects, she was frustrated by her inability to drive. After further discussion regarding potential adverse effects of increasing the dose of carbamazepine versus the benefits of achieving complete control of the seizures, a joint decision was made to increase the dose to 300 mg twice daily. However, a few weeks before her next follow-up appointment at 6 months, she telephoned stating that she was pregnant and wanted to know whether or not it would be safe to continue taking carbamazepine.
Treatment challenges
Anticonvulsant use and contraception: The hepatic enzyme-inducing anticonvulsants, especially carbamazepine (but also phenytoin, oxcarbazepine, phenobarbitone and, at doses > 200 mg daily, topiramate), reduce the efficacy of oral contraceptives by increasing their metabolism. Women who use anticonvulsants and oral contraception should be warned of potential contraceptive failure. Although higher-dose oestrogen preparations are often suggested, these are no guarantee, so it is more appropriate to consider other methods of contraception (eg, condoms, medroxyprogesterone with a 10-week cycle, intrauterine hormone-releasing systems). Alternatively, newer anticonvulsive drugs such as lamotrigine or levetiracetam may be more appropriate, as these largely avoid problems with hepatic metabolism and thus do not reduce the efficacy of oral contraception, although higher doses may be required for lamotrigine because oral contraception reduces serum lamotrigine levels.10
Anticonvulsant use in pregnancy: The management of epilepsy during pregnancy is a complex issue that should be undertaken by a neurologist who specialises in epilepsy. No anticonvulsant is completely safe. The older drugs are associated with an increased incidence of major congenital malformations, including cardiac, orofacial, urogenital, skeletal and neural tube defects. The risk appears to be highest with valproate, with evidence suggesting that it is best avoided if possible, especially during the first trimester (Grade B evidence),11 with doses above 800–1000 mg daily imparting the highest risk.12 Recent reports have noted cognitive deficits in some children born to mothers taking valproate, particularly at higher doses.13 Data regarding the newer drugs are insufficient for any clear recommendations to be made, but early data suggest they are comparable to the older agents.14 There is a broad consensus that carbamazepine is the safest drug to use in pregnancy (Grade B evidence).15,16
Uncontrolled epilepsy imparts a significant risk to the mother and baby. Although the effects of non-convulsive seizures on a developing fetus are not clear, convulsive episodes, accidents and falls are significant hazards. The recommended approach in pregnancy is treatment with the anticonvulsant drug that best controls seizures, aiming for monotherapy at the lowest possible dose. Changes to the medication regimen during pregnancy can be problematic due to exposure to polytherapy during the changeover period (the old drug should be withdrawn slowly and the new drug introduced slowly). If changes to the drug regimen are required, this is ideally done well before pregnancy to ensure adequate seizure control with the new treatment.11
Janice and her neurologist discussed the risks of anticonvulsant treatment versus no treatment during her pregnancy. Janice decided to remain on carbamazepine at a reduced dosage of 200 mg twice daily and was commenced on folate supplementation to minimise the risk of fetal neural tube defects. She was reminded that driving was not permitted until she had been seizure-free for 6 months.
Janice gave birth to a normal baby, after which her anticonvulsant medication was changed to levetiracetam (because she preferred to continue using oral contraception). She remained seizure-free and without any appreciable adverse effects, and her 3-monthly reviews were extended to 6-monthly and then 12-monthly. The importance of preconception planning for future pregnancies was reiterated.
Withdrawal of anticonvulsants: Medication withdrawal is a complex issue that should also be managed by a neurologist who specialises in epilepsy. The cumulative probability of remaining seizure-free 2 years after discontinuation of anticonvulsive medication has been estimated at 35%–57% for adults.17 Discontinuation may be a reasonable option for many patients who have been seizure-free for more than 2 years, particularly children. Many factors need to be considered, such as occupation and lifestyle, adverse medication effects, sex and driving status, and the risk of recurrent seizures compared with the risks of continuing anticonvulsant therapy.17 Predictors of a higher risk of seizure recurrence include partial seizures, known structural cerebral abnormality and abnormal EEG results, but none are absolute predictors and decisions about withdrawal of therapy must be individualised.
When a drug is withdrawn, it is important to give appropriate advice regarding safety, particularly with regard to driving, and unsupervised bathing and swimming. Generally, these activities should be avoided for the period of withdrawal and an additional 12 weeks, but advice must be individualised.
1 Differential diagnoses of unresponsive episodes and seizures that should be excluded before diagnosing focal epilepsy
Syncope should always be considered when there appears to be a specific precipitant, particularly pain, even if minor and considered to be irrelevant by the patient. Precipitants may include acute pain, anxiety, rising from the supine position, coughing and prolonged standing. There are usually one or more prodromal symptoms: nausea, lightheadedness, or tunnel or blurred vision. Onset is generally rapid, pallor and diaphoresis may be described by a witness, and brief convulsive motor activity and urinary incontinence are common features. The return to consciousness is usually rapid, but can be slower in older patients. Postictal confusion is not a feature, unless a significant head injury has occurred. If syncope is a diagnostic possibility, electrocardiography should be performed.
Generalised epilepsy usually has no warning, no preceding aura and no focal features. Motor activity is generalised from the outset in generalised tonic–clonic seizures.
Migraine involves progression of visual and/or sensory symptoms over a prolonged period, typically 15–20 minutes. Visual auras are common and tend to be linear or geometric. Motor activity is highly unusual. Migraine can occur without headache. Personal or family history of migraine should be sought.
Psychogenic non-epileptic seizures often last longer than epileptic seizures. The eyes are often closed (sometimes this is forced); motor behaviour may be asynchronous, with large-amplitude waxing and waning; and movements such as side-to-side rolling, head turning, thrashing of limbs and pelvic thrusting may be involved. Resolution of consciousness may be rapid without postictal confusion. These events occasionally coexist with epileptic seizures. An underlying psychiatric illness or history of psychological distress is not always present.
Panic attacks can occur in patients with an underlying anxiety or panic disorder. Attacks are of variable duration, but rarely last less than 5 minutes, and are often associated with palpitations, chest pain, hyperventilation and a sense of impending doom. They are not associated with altered awareness.
Tics are stereotyped movements or utterances with abrupt onset and brief duration. Premonitory feelings or sensations are relieved by execution of the tic. Tics may be temporarily suppressed voluntarily and are not associated with altered awareness.
Transient ischaemic attacks have an abrupt onset followed by full recovery within 24 hours. Unilateral hemiparesis, hemisensory loss or aphasia is common.
Transient global amnesia generally lasts longer than seizures, often up to several hours. It does not involve motor or sensory manifestations of seizures or other focal neurological signs, and has no postictal phase.
2 Janice’s electroencephalogram
 | |||||||||||||||
|
Abnormal sharp waves over the left temporal lobe are visible, with predominance in the anterior temporal region (red ovals). | |||||||||||||||
3 Coronal magnetic resonance imaging scan of Janice’s brain
 | |||||||||||||||
|
Atrophy of the left hippocampus with increased signal intensity (red circle) is visible in this fluid-attenuated inversion recovery (FLAIR) image. | |||||||||||||||
4 Drugs used to treat complex partial seizures
Competing interests
Mark Cook has received honoraria from UCB Pharma, sanofi-aventis and SciGen, and has undertaken clinical trials with PPD, PRA International and Eisai.
References
- Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010; 51: 676-685. 0_CHDEBGCD
- Chang BS, Lowenstein DH. Epilepsy. New Engl J Med 2003; 349: 1257-1266. 0_CHDGDDAH
- Perrig S, Jallon P. Is the first seizure truly epileptic? Epilepsia 2008; 49 Suppl 1: 2-7. 0_i1140536
- Krumbulz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review). Neurology 2007; 69: 1996-2007. 0_i1140538
- Glauser T, Ben-Menachem E, Bourgeois B, et al. ILAE treatment guidelines: evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006; 47: 1094-1120. 0_i1140540
- Marson AG, Williamson PR, Clough H, et al. Carbamazepine versus valproate monotherapy for epilepsy: a meta-analysis. Epilepsia 2002; 43: 505-513. 0_i1140542
- French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new onset epilepsy. Neurology 2004; 62: 1252-1260. 0_i1140544
- Sheth RD, Harden CL. Screening for bone health in epilepsy. Epilepsia 2007; 48 Suppl 9: 39-41. 0_i1140546
- Austroads. Assessing fitness to drive for commercial and private vehicle drivers. Medical standards for licensing and clinical management guidelines. Sydney: Austroads, 2003. 0_i1140548
- Zupanc ML. Antiepileptic drugs and hormonal contraceptives in adolescent women with epilepsy. Neurology 2006; 66 (Suppl 3): S37-S45. 0_i1140551
- Harden CL, Meador KJ, Pennell PD, et al. Management issues for women with epilepsy — focus on pregnancy (an evidence-based review): II. Teratogenesis and perinatal outcomes. Epilepsia 2009; 50: 1237-1246. 0_i1140553
- Vajda FJ, O’Brien TJ, Hitchcock A, et al. Critical relationship between sodium valproate dose and human teratogenicity: results of the Australian register of anti-epileptic drugs in pregnancy. J Clin Neurosci 2004; 11: 854-858. 0_i1140555
- Meador KJ, Baker GA, Browning N, et al. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N Engl J Med 2009; 360: 1597-1605. 0_i1140557
- Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA 2011; 305: 1996-2002. 0_i1140559
- Tomson T, Battino D, Bonizzoni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol 2011; 10: 609-617. 0_i1140561
- Vajda F. The Australian Pregnancy Register of Anti-epileptic Drugs: 10 years of progress. J Clin Neurosci 2010; 17: 1485-1488. 0_i1140563
- Chadwick D. Starting and stopping treatment for seizures and epilepsy. Epilepsia 2006; 47 Suppl 1: 58-61. 0_i1140565
Provenance: Commissioned; externally peer reviewed.