





Volume 195 - Issue 10
MELAS syndrome in an Indigenous Australian woman
Authors: Luke J Conway, Thomas E Robertson, James J McGill and Josh P Hanson
Med J Aust 2011; 195 (10): 581-582. || doi: 10.5694/mja10.11393
Published online: 21 November 2011
Published online: 21 November 2011
To the Editor: MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes) syndrome has not been reported previously in the Aboriginal Australian population. Here, we describe a patient with MELAS syndrome in this population.
A 29-year-old Aboriginal Australian woman presented with a 3-day history of seizures and confusion and a background of cognitive impairment, sensorineural deafness, epilepsy and short stature. On admission, she weighed 29.9 kg and was 1.46 m tall (body mass index, 14 kg/m2). She had myopathic facies, generalised mild motor weakness (4/5) and brisk reflexes (3+). These neurological findings represented a stepwise deterioration from previous assessments.
On admission, the patient’s blood count, liver function tests, and serum urea, electrolytes and creatinine levels were normal. Her plasma bicarbonate level, anion gap and thyroid function were also normal.
The patient had elevated resting arterial lactate (2.7 mmol/L; reference interval [RI], 0.7–2.5 mmol/L) and pyruvate (98 μmol/L; RI, 30–90 μmol/L) levels. The cerebrospinal fluid (CSF) protein concentration was 820 mg/L (RI, 150–500 mg/L) and the CSF lactate level was 5.4 mmol/L (RI, 0.7–2.5 mmol/L).
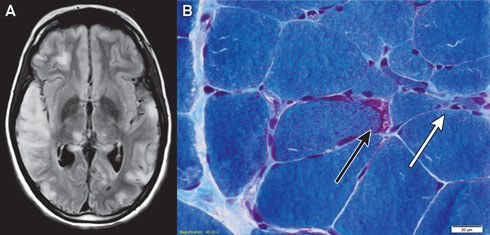
Magnetic resonance imaging of the patient’s brain demonstrated increased T2 signal involving grey and white matter throughout both cerebral hemispheres, most confluent in the right temporal and parietal lobes (Box 1, A). An electroencephalogram displayed a slow rhythm with epileptic activity in the temporal and centroparietal regions.
Histological examination of a biopsy of the gastrocnemius muscle was consistent with MELAS syndrome (Box 1, B). Electron microscopy of a muscle biopsy sample revealed mitochondria with abnormally arranged cristae and abnormal electron densities. Mitochondrial respiratory chain enzyme studies on muscle samples were within normal limits, but the common m.3243A>G mutation in the MTTL1 gene was detected in about 70% of the mitochondrial DNA (mtDNA) in muscle tissue and in 10% of the mtDNA of the peripheral blood.
MELAS syndrome is a maternally inherited multisystem disorder resulting from mutations in mtDNA.1 Mitochondrial dysfunction leads to the clinical phenotype and lactic acidosis. Cerebral ischaemia unrelated to vascular territories is suggestive of the diagnosis,2 which is confirmed by genetic studies, enzyme assays and histological examination of affected tissue.3
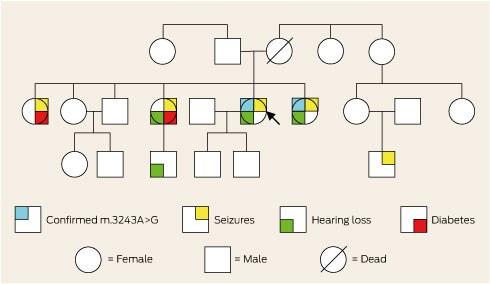
The patient’s family was of Aboriginal Australian descent and she was not aware of any European ancestry. Further inquiry revealed a family history of deafness, diabetes and epilepsy (Box 2). Consequently, the family were offered genetic counselling. A literature review prompted initiation of arginine and coenzyme Q10 therapy.3 Twelve months later, she had good seizure control and had not been re-hospitalised.
The disproportionate differences in health between Indigenous and non-Indigenous Australians are often appropriately ascribed to environmental factors. However, potentially treatable causes should always be considered.
Competing interests
No relevant disclosures.
References
- Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalo-myopathies. Nature 1990; 348: 651-653. 0_BHGHCFAG
- Suzuki T, Koizumi J, Shiraishi H, et al. Mitochondrial encephalomyopathy (MELAS) with mental disorder. CT, MRI and SPECT findings. Neuroradiology 1990; 32: 74-76. 0_i1142886
- Santa KM. Treatment options for mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. Pharmacotherapy 2010; 30: 1179-1196. 0_CBBJIEJA