





Volume 194 - Issue 4 Supplement
Advances in structural and molecular neuroimaging in Alzheimer’s disease
Authors: Kathryn A Ellis, Christopher C Rowe, Cassandra E I Szoeke, Victor L Villemagne, David Ames, Gaël Chételat, Ralph N Martins, Colin L Masters, Jurgen Fripp, Oscar Acosta, Parnesh Raniga, Pierrick T Bourgeat and Olivier Salvado
Med J Aust 2011; 194 (4 Suppl): S20. || doi: 10.5694/j.1326-5377.2011.tb02938.x
Published online: 21 February 2011
Published online: 21 February 2011
Abstract
Longer life expectancies lead to increases in the prevalence of age-associated illnesses. The number of Australians with dementia is predicted to rise, from 234 000 in 2009 to over 1 million by 2050, as a result of the increased prevalence of Alzheimer’s disease (AD), the leading cause of dementia in the elderly.
Early diagnosis of AD will become more important as disease-modifying therapies emerge within the next decade.
Advances in molecular neuroimaging with amyloid-β-specific radioligands for positron emission tomography, aided by magnetic resonance imaging techniques, allow detection of AD years before symptoms of dementia develop.
Longitudinal prospective studies, such as the Australian Imaging Biomarkers and Lifestyle (AIBL) study of ageing, will determine the sensitivity and specificity of these analysis techniques for diagnosing AD and predicting cognitive decline.
Longer life expectancies are accompanied by increases in the prevalence of illnesses associated with ageing. As age is the major risk factor for Alzheimer’s disease (AD) — the leading cause of dementia in the elderly — the prevalence of AD is predicted to increase, with the number of Australians with dementia rising from 234 000 in 2009 to over 1 million by 2050 (ie, from 1% to 3% of the Australian population).1 Such increases in dementia prevalence will have major consequences for the Australian health care system.1 On the other hand, if the average onset of AD could be delayed by 5 years, there would be a 50% reduction in new cases each year.2
AD is a progressive neurodegenerative disorder, clinically characterised by cognitive decline, and histologically identified by the presence of amyloid-β (Aβ) plaques and neurofibrillary tangles in the brain.3 The formation and deposition of Aβ in the brain are central to current aetiological theories of AD.4 The Aβ theory of AD is that toxicity associated with accumulation of Aβ leads to synaptic and neuronal degeneration and subsequent grey-matter atrophy.3 There is considerable evidence to suggest that Aβ accumulation begins several years before neuronal death and up to decades before symptoms of cognitive decline develop.5
Since the 1970s, neuroimaging has been routinely used in the clinical evaluation of patients with suspected AD.6 Traditionally, structural imaging has been applied to exclude or evaluate potentially treatable causes of cognitive impairment, such as multiple small strokes and normal pressure hydrocephalus. More recently, patterns of atrophy have been used to assist in diagnosing dementia subtypes (eg, hippocampal atrophy to support a diagnosis of AD). In clinical practice, structural scans are generally interpreted visually. The qualitative nature of this approach means that much of the richness of the data is not exploited. However, new technologies are being developed that can significantly increase the yield from each scan.
Perhaps the most exciting advance in neuroimaging of AD during the past 10 years is the development of Aβ-imaging agents for positron emission tomography (PET) that can detect Aβ in the brain many years before atrophy occurs or clinical symptoms manifest themselves.7,8 Furthermore, the capacity to combine structural and molecular imaging technologies is generating results with greater diagnostic sensitivity and specificity, and indications are that using these techniques will allow earlier diagnosis of AD in clinical practice.
Studies such as the Australian Imaging Biomarkers and Lifestyle (AIBL) study of ageing, initiated by the CSIRO (Commonwealth Scientific and Industrial Research Organisation), have assembled large cohorts of individuals who can be assessed, compared and followed up over a long period of time to facilitate prospective research into ageing and AD. The AIBL study is the largest cohort study of its kind in Australia (and one of the largest worldwide) to thoroughly assess individuals with and without AD and at varying levels of risk of developing AD.9 A key component of the AIBL study was conducting high-quality magnetic resonance imaging (MRI) and in-vivo PET imaging of Aβ in the brain in a substantial proportion of the cohort.
Here, we provide an overview of the typical MRI and PET imaging characteristics, and we discuss the contribution of the AIBL study and enhanced image analysis to more accurate and earlier diagnosis of AD.
Structural imaging in dementia — current and future use
Structural imaging techniques are routinely used to identify and diagnose potentially treatable causes of cognitive impairment.10,11 Medial temporal-lobe atrophy, including the hippocampus and entorhinal cortex, is an early characteristic feature of AD, and hippocampal atrophy is strongly associated with memory decline.11
Structural images provide significantly more information when processed with automatic image analysis software. The rate of hippocampal atrophy is two to three times greater in early AD than with normal ageing,11 but changes are small and require precise measurement.
Two recent technologies have emerged that greatly facilitate detailed MRI analysis. The first is automatic segmentation of the brain into its three main tissues — white matter, grey matter and cerebrospinal fluid — using intensity-based tissue classification. The second is the ability to spatially align or warp an individual brain scan to a template on which anatomical structures have been defined. As a result, volume (eg, hippocampus, ventricles) or thickness (eg, cortex) can be automatically estimated at the voxel level, or for given regions of interest (eg, lobes or Broca’s area). (A voxel or volumetric pixel is a volume element, representing a value on a regular grid in three-dimensional space.) Statistical analysis can then be conducted in a large cohort, such as that in the AIBL study, to identify atrophy patterns,12,13 but also to estimate atrophy in individual patients and compare with atrophy patterns corrected for confounding factors (eg, age, sex, years of education). These techniques have begun to enter clinical practice, but are not routinely available in Australia.
Aβ imaging — a new paradigm emerges
Aβ imaging enables in-vivo detection and quantitative estimation of levels of Aβ in the brain,7,8,14 and is considered a putative biomarker for AD. The most studied and as such validated Aβ imaging ligand is N-methyl [11C]2-(4‘-methylaminophenyl)-6-hydroxybenzothiazole, or Pittsburgh compound B (PiB). PiB binds specifically to fibrillar Aβ deposits in brain tissue obtained at autopsy, and a high correlation of regional PiB binding to plaque load has been found at autopsy in a patient who had previously undergone PiB-PET imaging.15 PiB is regarded as a sensitive marker for Aβ in healthy older individuals with apparently normal cognition, in those with mild objective memory impairment, and in patients with AD.8,16,17
PiB-PET images show relatively low, non-specific, homogeneous PiB retention in white matter, while the variable retention in grey matter reflects fibrillar Aβ burden (Box). In patients with AD, high PiB retention can be readily visualised in the cortical mantle on inspection of images and, for the purposes of early detection and monitoring of progression, the advantages of quantitative measurements of Aβ deposition have been demonstrated.17 However, quantitative estimation of PiB binding is challenging and care must be taken to process the data correctly.
The first step is normalisation of the PiB signal to the signal in the cerebellar cortex, where PiB binding has been shown to be very low, reflecting an absence of dense Aβ deposition in that area. The resulting parameter is called the standardised uptake value ratio.18 The second important challenge is associated with the limited resolution of PET scans, which results in three issues when quantifying PiB images.
First, current PET camera resolution is low (~ 6 mm) compared with the thickness of the cortex (~ 3 mm), resulting in blurring or partial volume effect.19,20
Second, the retention in grey matter can be lower (for healthy individuals) or higher (for subjects with AD) than in white matter, confounding signal measurement in the cortex due to variable spillover into and from surrounding white matter and cerebrospinal fluid.
Third, atrophy associated with AD has the effect of reducing PiB signal intensity, potentially masking an increase in PiB retention per volume of grey matter.
Similar to atrophy patterns, PiB retention patterns can be computed, allowing a quantitative estimation of Aβ deposition at the voxel or regional level. Recent improvement in these techniques has revealed differences between grey matter atrophy and Aβ deposition patterns.20 Voxel-wise and regional correlation between PiB binding, atrophy, and other clinical measurements (eg, Mini-Mental State Examination score) have identified few relationships and only in specific brain areas. For example, the AIBL study has shown that hippocampal atrophy is correlated with PiB retention in the inferior temporal lobe only and not elsewhere, and only at the earliest stages of the disease in healthy, asymptomatic individuals with high PiB binding.13,21 However, it has also become clear from our studies that, while Aβ must be present for a diagnosis of AD to be made and it appears before atrophy and cognitive decline, there is not a direct, robust dose-dependent relationship between Aβ and cross-sectional measures of atrophy or measures of cognition, suggesting that other “downstream” factors are the predominant determinants of neurodegeneration.17,22 A more definitive understanding of the relationship of Aβ to neurodegeneration will emerge from the longitudinal findings of the AIBL study, which is currently in its third year of follow-up.
Implications for the routine diagnosis of AD
Application of these advanced imaging and analysis techniques to the AIBL study population has revealed a high prevalence of Aβ deposition in the brain with ageing. The prevalence of Aβ deposition increases rapidly with age above 60 years, such that, by the late 80s, the prevalence of cognitively unimpaired subjects with high PiB retention in the brain exceeds 60%.23
The AIBL study has confirmed that genetic factors play a large role in the appearance of Aβ. The presence of at least one copy of the apolipoprotein E (APOE) ε4 gene allele is associated with both earlier and greater deposition of Aβ in the brain. PiB scans show Aβ deposition in 50% of asymptomatic elderly carriers of the APOE ε4 allele compared with only 18% of non-carriers.23 Furthermore, the AIBL study has shown that participants with mild objective memory impairment who have deposition of Aβ in the brain, as shown by a positive PiB scan, are at much greater risk of AD over the following 2 years (60%) than those with a negative PiB scan (< 10%).22 Over 90% of individuals with a clinical diagnosis of AD have positive results of a PiB scan, while patients with frontotemporal dementia show negative results.8,14,23
Correlation of structural changes that reflect synaptic and neuronal loss with Aβ imaging has confirmed that Aβ deposition precedes loss of brain cells.23 Our studies have also confirmed that Aβ accumulation is a very slow process, with levels increasing by 2%–3% a year, implying that cognitive decline follows Aβ deposition by many years.22 These imaging-based observations support the proposal that Aβ accumulation begins several years before neuronal death and up to decades before cognitive decline.5
Fluorine-18-labelled PET radiotracers for imaging cerebral Aβ deposition are being studied in a Phase III trial and are likely to be available for clinical use within 2 years.24-26 These radiopharmaceuticals can be mass produced and widely distributed, unlike the carbon-11-labelled compounds currently used in research settings, which have a very short half-life.
Progress in early diagnosis of AD using advanced neuroimaging techniques is currently outpacing the development of effective disease-modifying therapy, leading to questions being asked about the value of early diagnosis and these expensive technologies. However, imaging and analysis techniques provide a way of detecting AD before the onset of significant cognitive and functional decline, and therefore offer the potential for intervention to prevent deterioration with novel therapeutic approaches. Indeed, it is likely that safe and effective disease-modifying therapies for AD will emerge within the next decade,27 making early diagnosis of AD critically important. Furthermore, the longitudinal prospective design of studies, such as the AIBL study, will allow the determination of the sensitivity and specificity of these analysis techniques for accurate diagnosis of AD, and, more importantly, the elucidation of their predictive value for cognitive decline.
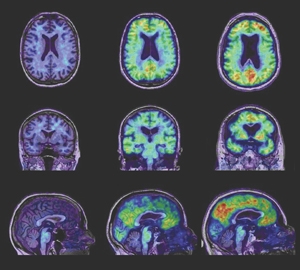
In-vivo imaging of burden of amyloid-β in ageing and dementia: comparison of brain scan images — healthy control (column 1), mild cognitive impairment (column 2), and Alzheimer’s disease (column 3)
Representative transaxial (top row), coronal (middle row) and sagittal (bottom row) images from positron emission tomography / magnetic resonance imaging of a 76-year-old healthy control subject (HC [column 1] — Mini-Mental State Examination [MMSE] score, 30), a 76-year-old subject with mild cognitive impairment (MCI [column 2] — MMSE score, 28), and a 75-year-old patient with Alzheimer’s disease (AD [column 3] — MMSE score, 25). The images show clear differences when comparing the HC subject and the patient with AD, with non-specific Pittsburgh compound B (PiB) binding in white matter in the HC subject compared with PiB binding in the frontal, temporal and posterior cingulate / precuneus cortex of the patient with AD and marked atrophy. The subject with MCI presents similar patterns of cortical atrophy and PiB binding to those of the patient with AD, but to a lesser degree.
Competing interests
Cassandra Szoeke has received honoraria, to 2009, from Pfizer and Lundbeck and, to 2006, from Sanofi-Aventis and Mayne Pharma. David Ames has received remuneration for board membership of Pfizer, Novartis and Janssen-Cilag, and travel expenses from Novartis (2009).
Acknowledgements
Core funding for the AIBL study was provided by the CSIRO and was matched by contributions from the study partners (see http://www.aibl.csiro.au). The study also receives support from the National Health and Medical Research Council and the Dementia Collaborative Research Centres program (DCRC2). Pfizer International has contributed financial support to assist with analysis of blood samples and to further the AIBL research program. The Alzheimer’s Association (USA) has contributed support to allow AIBL neuroimaging stream data (and accompanying clinical and cognitive data) to be made available via the Alzheimer’s Disease Neuroimaging Initiative website. We thank Alzheimer’s Australia (Victoria and Western Australia) who have assisted with promotion of the study and screening of telephone calls from volunteers, and collaborated with AIBL volunteer functions. Cassandra Szoeke is partially supported by a research fellowship funded by Alzheimer’s Australia. The AIBL team wishes to thank all those who took part as subjects in the study for their commitment and dedication to helping advance research into the early detection and causation of AD.
References
- Access Economics. Keeping dementia front of mind: incidence and prevalence 2009–2050. Canberra: Alzheimer’s Australia, 2009. 0_BABJHFCH
- Access Economics. Delaying the onset of Alzheimer’s disease: projections and issues. Canberra: Alzheimer’s Australia, 2004. 0_BABHDJJH
- Masters CL, Cappai R, Barnham KJ, et al. Molecular mechanisms for Alzheimer’s disease: implications for neuroimaging and therapeutics. J Neurochem 2006; 97: 1700-1725. 0_i1096021
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med 2010; 362: 329-344. 0_i1096023
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297: 353-356. 0_i1096025
- Ames D, Chiu E, editors. Neuroimaging and the psychiatry of late life. Cambridge: Cambridge University Press, 1997: 101. 0_i1096027
- Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 2004; 55: 306-319. 0_i1096029
- Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007; 68: 1718-1725. 0_i1096031
- Ellis KA, Bush AI, Darby D, et al. The Australian Imaging, Biomarkers and Lifestyle flagship study of aging (AIBL): methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr 2009; 21: 672-687. 0_i1096033
- Whitwell JL, Jack CR Jr. Neuroimaging in dementia. Neurol Clin 2007; 25: 843-857. 0_i1096035
- Scheltens P, Fox N, Barkhof F, et al. Structural magnetic resonance imaging in the practical assessment of dementia: beyond exclusion. Lancet Neurol 2002; 1: 13-21. 0_i1096037
- Acosta O, Bourgeat P, Zuluaga MA, et al. Automated voxel-based 3D cortical thickness measurement in a combined Lagrangian-Eulerian PDE approach using partial volume maps. Med Image Anal 2009; 13: 730-743. 0_i1096039
- Bourgeat P, Chételat G, Villemagne VL, et al. β-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology 2010; 74: 121-127. 0_i1096041
- Rabinovici GD, Jagust WJ. Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav Neurol 2009; 21: 117-128. 0_i1096043
- Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 2008; 131: 1630-1645. 0_i1096045
- Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 2008; 65: 1509-1517. 0_i1096047
- Villemagne VL, Fodero-Tavoletti MT, Pike KE, et al. The ART of loss: Aβ imaging in the evaluation of Alzheimer’s disease and other dementias. Mol Neurobiol 2008; 38: 1-15. 0_i1096049
- Raniga P, Bourgeat P, Fripp J, et al. Automated 11C-PiB standardized uptake value ratio. Acad Radiol 2008; 15: 1376-1389. 0_i1096051
- Soret M, Bacharach SL, Buvat I. Partial-volume effect in PET tumor imaging. J Nucl Med 2007; 48: 932-945. 0_i1096053
- Fripp J, Bourgeat P, Acosta O, et al. Appearance modelling of C11 PiB PET images: characterizing amyloid deposition in Alzheimer’s disease, mild cognitive impairment and healthy aging. Neuroimage 2008; 43: 430-439. 0_i1096055
- Chételat G, Villemagne VL, Bourgeat P, et al. Relationship between atrophy and β-amyloid deposition in Alzheimer’s disease. Ann Neurol 2010; 67: 317-324. 0_i1096057
- Villemagne V, Pike KE, Chételat G, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer’s disease. Ann Neurol 2011; 69: 181-192. 0_i1096059
- Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010; 31: 1275-1283. 0_i1096061
- Vandenberghe R, Laere K, Ivanoiu A, et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol 2010; 68: 319-329. 0_i1096063
- Wong D, Rosenberg PB, Zhou Y, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir F 18). J Nucl Med 2010; 51: 913-920. 0_i1096066
- Rowe CC, Ackerman U, Browne W, et al. Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol 2008: 7: 129-135. 0_i1096067
- Ritchie CW, Ames D, Masters CL, Cummings J. Therapeutic strategies in dementia. Oxford: Clinical Publishing, 2007: xiii. 0_i1096069
Motor Neuron Disease Mortality Trends in Australia From 1986 to 2023: A Population-Based Study
Carol M. Y. Lee, Rupendra N. Shrestha, Julian Gold, Mark Stevenson, Kelly L. Williams, Lyndal Henden, Sandrine K. K. Chan Moi Fat, Zoe Zussa, Amanda Wright, Martin Jude, Mark A. S. Laidlaw, Willem J. R. Fokkink, Dominic B. Rowe
Mild traumatic brain injury and concussion and persisting post‐concussion symptoms: new guidelines to support evidence‐based assessment and management in Australia and Aotearoa New Zealand
Karen M Barlow, Jennie L Ponsford, Alice Theodom, Gill Cowen, Gavin A Davis, Vicki Anderson, Franz E Babl, David Cole, Jennifer Cullen, Stuart R Dalziel, Melinda Fitzgerald, Howard Flavell, Caroline Yates, Rebecca Kimble, John H Olver, Rhonda Orr, Mark Ralfe, Michael Rose, Nick Rushworth, Julia Treleaven, Gary Browne, Nathan Delang, Sarah Harris, Gary Mitchell, Sean Tweedy
The prevalence of and variation in indicators of the quality and safety of long term aged care in Australia, 2019: a cross‐sectional population‐based study
Tesfahun C Eshetie, Gillian E Caughey, Catherine Lang, Olivia Ryan, Renuka Visvanathan, Craig Whitehead, Keith Evans, Janet K Sluggett, Jyoti Khadka, Carolyn Dawkins, Helena Williams, Miranda Starke, Sara Blunt, Anne Liddell, Megan Corlis, Anna Sheppeard, Penelope Lello, Marilyn Thien, Steven L Wesselingh, Maria C Inacio
Health and economic benefits of improving pre‐hospital identification of stroke in Australian women: a modelling study
Thomas Gadsden, Lei Si, Emily R Atkins, Cheryl Carcel, Xia Wang, Stephen Jan, Mark Woodward, Laura E Downey
Symptom burden, quality of life, and diagnostic journey of people with postural orthostatic tachycardia syndrome, Australia, 2021–24: a descriptive patient registry data study
Marie‐Claire Seeley, Gemma Wilson, Eric Ong, Amy Langdon, Jonathan Chieng, Danielle Bailey, Kristina Comacchio, Amanda Page, Dennis H Lau, Celine Gallagher
The intersection of rurality and dementia prevalence in Australia for Aboriginal and Torres Strait Islander and non‐Indigenous peoples
Antonia J Clarke, Marwan Ahmed, Judith M Katzenellenbogen, John Towney (Wiradjuri), Anna H Balabanski, Adrienne Withall (Dharawal Yuin), Kylie Radford, Amy Brodtmann