





Volume 194 - Issue 1
The prevalence and diagnosis rates of Klinefelter syndrome: an Australian comparison
Authors: Amy S Herlihy, Jane L Halliday, Megan L Cock and Robert I McLachlan
Med J Aust 2011; 194 (1): 24-28. || doi: 10.5694/j.1326-5377.2011.tb04141.x
Published online: 3 January 2011
Published online: 3 January 2011
Abstract
Objective: To determine the prevalence and diagnosis rates of Klinefelter syndrome (KS) in Victoria, Australia, and compare these to previous international findings.
Design, setting and participants: A Victorian population-based descriptive study of all cytogenetic examinations resulting in a diagnosis of KS, including prenatal diagnoses from 1986 to 2006 and postnatal diagnoses from 1991 to 2006.
Main outcome measures: Birth prevalence and diagnosis rates of KS.
Results: The birth prevalence of KS in Victoria is estimated to be 223 per 100 000 males (95% CI, 195–254), with about 50% of cases remaining undiagnosed.
Conclusions: KS may be occurring more frequently than has been reported previously, yet many cases remain undiagnosed. Our results highlight the need for increased awareness leading to timely detection.
Klinefelter syndrome (KS) is a genetic condition affecting males that is most often caused by an additional X chromosome (47,XXY karyotype).1 This results in a spectrum of features ranging from azoospermia, small testes, androgen deficiency and gynaecomastia, to varying degrees of learning and behavioural difficulties2 (Box 1). The number and severity of these features are known to be highly variable between individuals.
A mean prevalence for KS of 152 per 100 000 male births was estimated from newborn screening programs in the 1960s and 1970s in several countries, including Denmark, the United States, Canada, Japan and the United Kingdom.3-10 Despite this high frequency, and features such as small testicles in adulthood, it has been estimated that less than 10% of the estimated number of affected fetuses are detected prenatally, and only 26% of live-born cases are diagnosed postnatally.11 A birth prevalence for KS of 153 per 100 000 males in Denmark has been estimated using population information and adjusting the prenatal prevalence for maternal age, as KS is an incidental finding of prenatal karyotype tests that are more commonly performed in older mothers.10 Comparison with postnatal diagnoses confirmed that only 25% of KS cases are detected.
The low diagnosis rate suggests most males with KS will not receive potentially beneficial treatments, especially androgen therapy.2 Most adult diagnoses occur during fertility assessment, beyond the ideal point for intervention.10 Detection in childhood and timely intervention may be essential for optimal medical and psychosocial outcomes in adulthood.12 Underdiagnosis may be due to men’s hesitancy about seeking medical attention, low awareness of KS among health professionals,13 and failure by health professionals to perform routine genital examinations in adult men.
A recent study suggests that the prevalence of KS may be increasing.14 The 47,XXY karyotype is almost always the result of meiotic non-disjunction during parental gamete formation, which increases with both maternal and paternal age.15,16 Given increasing parental age trends in Australia,17 and the potential clinical significance of non-diagnosis of KS, it is imperative to establish current prevalence estimates. We have used population data from Victoria to estimate local prevalence and diagnosis rates of KS.
Methods
Karyotypes
Any karyotype including a single Y chromosome and more than one X chromosome, where the individual was listed as a male, was included in our analysis. Mosaic karyotypes were also included, unless a female 46,XX line was present.
Data sources
General population data, including birth numbers and maternal ages, were collected from the following sources.
Births in Victoria — a report on notifications of births at 20 weeks’ gestation and beyond, for which reporting is mandatory, that has been produced by the Victorian Perinatal Data Collection Unit (VPDCU) since 1983 and contains comprehensive information on mothers and babies.18
Australian Bureau of Statistics birth and population data by age, sex and state.19
Data on diagnoses of KS in Victoria were collected from all relevant sources.
The Victorian Birth Defects Register (VBDR) — a population-based surveillance system held by the VPDCU, active since 1983. Birth defects (including sex chromosome anomalies) diagnosed prenatally and postnatally up to 16 years of age are reported to the VBDR, and data were available up to and including 2005.
The Victorian Prenatal Diagnosis Database (VPDD), which contains data from cytogenetic laboratories on every chorionic villus sampling and amniocentesis procedure performed in Victoria, including indications for diagnostic testing, karyotypes and maternal variables. Data on prenatal diagnoses of KS were available for the period 1986–2006.
Two public and two private cytogenetic laboratories that undertake all karyotype testing in Victoria. These four laboratories provided data on prenatal diagnoses of KS for the period 1987–2006 and on postnatal diagnoses for 1991–2006.
We combined data for males diagnosed with KS into a single database and removed duplicates. The ages of mothers at the time of prenatal diagnostic testing and males at postnatal diagnosis were calculated based on dates of birth and dates of tests. Data were de-identified, and the final dataset included prenatal diagnoses for the period 1986–2006 and postnatal diagnoses for 1991–2006.
Statistical analyses
The prenatal prevalence of KS per 100 000 males tested for the period 1986–2006 was calculated by multiplying the number of prenatal diagnoses of KS in each year by 100 000, then dividing by the estimated number of prenatal tests carried out on male fetuses in each year.
Birth prevalence was estimated by adjusting the prenatal prevalence for maternal age using direct standardisation. Maternal age-specific rates of pregnancies involving a fetus with KS for the period 1986–2006 were calculated for each specified age group by dividing the number of prenatal diagnoses by the number of mothers undergoing prenatal diagnostic testing in each age group. This age-specific rate was applied to the population of women giving birth in each age group to estimate the expected number of births of babies with KS.
Age-specific, average annual postnatal diagnosis rates for the period 1991–2006 were calculated from the total number of cases diagnosed and the average annual population of Victoria in each 5-year age group from 0–4 years to 80–84 years over that period. The cumulative postnatal diagnosis rate (an estimate of cumulative diagnosis probability) from the beginning to the end of each 5-year age group was then calculated as five times the average annual postnatal diagnosis rate for that age group. Therefore, the cumulative diagnosis rate from birth to the end of any age group is the sum of age-specific cumulative diagnosis rates to that age. Postnatal diagnosis rates for the first and second 8-year periods of the study were compared.
The proportion of cases being diagnosed prenatally was estimated by taking the number of prenatal diagnoses of KS as a proportion of the expected number of cases of KS, based on the calculated birth prevalence rate. The proportion of all cases being diagnosed postnatally was estimated by taking the cumulative postnatal diagnosis rate of KS to age 84 as a proportion of the estimated birth prevalence.
Data were analysed using StataCorp statistical software, version 10, 2007 (StataCorp, College Station, Tex, USA). Binomial distribution for proportions, and Poisson distribution for rates were used to estimate 95% confidence intervals.
Results
Karyotypes
The numbers of each KS karyotype identified prenatally and postnatally are shown in Box 2. The overall proportion of KS karyotypes other than 47,XXY was 14.2%. The proportion of prenatal diagnoses with non-47,XXY KS karyotypes was 20.4%, while for postnatal diagnoses it was lower, at 12.3% (P < 0.01).
Prenatal prevalence
A total of 152 fetuses were identified as having a KS karyotype out of 85 650 tested between 1986 and 2006. As male fetuses make up about 51% of births,17 the denominator for prevalence calculations is 43 682 (Box 3). This resulted in a crude prevalence of 348 per 100 000 males tested (95% CI, 295–408).
Birth prevalence
The mean age of Victorian women having prenatal diagnostic testing was 36.4 years in 1986 and 35.9 years in 2006 (average across the study period, 36.0 years), and the mean age of Victorian women giving birth was 27.6 years in 1986 and 30.6 years in 2006 (average across the study period, 29.6 years). Direct maternal age standardisation of the prenatal prevalence resulted in an adjusted birth prevalence of 223 per 100 000 male births (95% CI, 195–254) (Box 4).
Postnatal diagnosis rate
A total of 487 cases of KS were diagnosed postnatally between 1991 and 2006 (Box 5). The cumulative postnatal diagnosis rate of KS to age 84 was estimated at 87 per 100 000 males (95% CI, 70–107). There was no significant difference between postnatal diagnosis rates for the first and second 8-year periods within the study (85 v 95 per 100 000 males; P < 0.46).
Overall diagnosis rate
Between 1986 and 2006, 675 439 mothers gave birth to 685 418 boys in Victoria. Based on the birth prevalence of 223 per 100 000 males, 1528 births with KS would be expected in the time period. As 152 cases of KS were diagnosed prenatally during this time, about 9.9% of expected cases of KS were diagnosed prenatally.
The cumulative postnatal diagnosis rate of 87 per 100 000 males provided an estimate of 39% as the proportion of expected births of babies with KS diagnosed postnatally.
Therefore, the addition of prenatal and postnatal diagnosis rates gives an estimated 49% of all expected cases of KS being diagnosed, with around 51% of males who have KS remaining undiagnosed in Victoria.
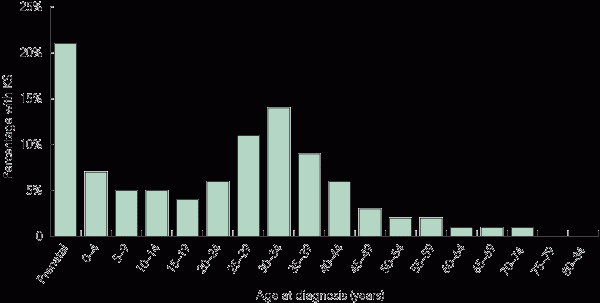
Age at diagnosis
Box 6 shows that, among the detected cases of KS, most were diagnosed prenatally (21.2%) or at 30–34 years of age (14.2%). A decline in diagnoses was seen from the age of 35 years, with few men being diagnosed beyond 50 years of age.
Discussion
Our estimates of the birth prevalence of KS in Victoria for the period 1986–2006 and the proportion of expected cases of KS that were diagnosed indicate that KS is both common and underdiagnosed. They also provide an indication of the prevalence of KS across Australia, which may be 25 000 cases in total, with almost 13 000 remaining undiagnosed.
The Victorian birth prevalence rate is higher than that seen in Denmark, but falls within the range of 85 to 223 per 100 000 male births seen among the combined newborn screening surveys carried out in several countries.3-9 One factor contributing to this higher rate may be the difference in proportions of karyotypes other than 47,XXY: 14.2% in Victoria compared with 10.2% from the Danish data. However, a sensitivity analysis (data not shown) showed that, even assuming a proportion of non-47,XXY karyotypes similar to Denmark, the birth prevalence in Victoria would still be higher (194 v 153 per 100 000 males; P < 0.028). There was a significant difference (P < 0.01) between prenatal and postnatal diagnoses of these other karyotypes, possibly because live-born males with a mosaic karyotype experience fewer clinical symptoms and escape detection more often than 47,XXY males, while all karyotypes have equal likelihood of prenatal detection.
Recently, a high prevalence of KS was found in an Asian cohort (355 per 100 000 males).20 Given that more than 8% of the Australian population is of Asian descent,19 compared with 3% of the Danish population,21 this may be an additional contributing factor to the higher birth prevalence of KS seen in Victoria. Unfortunately, the ethnic backgrounds of males with KS in our dataset were not available.
Another contributing factor to the higher prevalence of KS in Victoria may be maternal age, not only at the time of prenatal diagnostic testing, but also at the time of birth. The average age of Victorian women having prenatal testing is 36.0 years, compared with 34.0 years for the Danish population,10 and these differences have been adjusted for by standardisation. However, at the population level, Victorian women giving birth are, on average, older than Danish women (29.6 years v 28.1 years for each study period). While this age difference is not large, variations between maternal age distributions may have a significant impact on the number of KS cases.
In addition, paternal age is increasing. In Australia, the average age of fathers reached an all time high of 33.1 years in 2006, with the number of men having children in their 50s increasing by around 20% over the past decade.17
As well as a higher birth prevalence for KS, our calculations indicate a higher postnatal detection rate than that seen in Denmark10 or the UK.11 Our estimates are based on a more recent study period (1991–2006) than the Danish study (1970–2000),10 during which there has been consistent growth in access to fertility investigations in Australia.22 This may partly account for the higher detection rate and is consistent with the finding that most men are diagnosed between 25 and 39 years of age — the peak reproductive period.
Data strengths and limitations
We ensured that our data were complete by cross-checking multiple sources. To account for any gaps in prenatal reporting to the VBDR, we also collected data from the VPDD. Data were requested from cytogenetic laboratories to capture cases of KS not detected until adulthood. Complete postnatal data were only available for the period 1991–2006.
The postnatal diagnosis rate assumes that detection of KS has remained relatively constant over time, and that detection will occur before 85 years of age. As rates did not vary significantly between the first and second halves of the study period, the overall rate calculated here is likely to be a good indicator of current diagnosis rates. Finally, small numbers of KS diagnoses in younger maternal age groups suggest that our age-specific rates are most accurate for maternal ages of 35 years or higher.
Clinical and policy implications
The health outcomes consequent on making the diagnosis of KS are unclear, but an array of biomedical and psychosocial endpoints can be identified for which there are both empirical and evidence-based data in support of interventions. For example, up to 85% of males with KS will be testosterone-deficient after puberty,2 which may have profound medical and psychosocial impacts. Treatment may alleviate the symptoms,23 in addition to reducing condition-related morbidity and mortality.24
That most men with KS remain undiagnosed and “invisible” to the health system presents a number of challenges: for professional education, in terms of raising awareness and normalising routine genital examinations; for public health strategy, in terms of consideration of routine screening; and for research, in terms of risks and benefits of various interventions across the lifespan.
A number of important questions remain unanswered. Are men with undetected KS free from significant health problems, and do they therefore reasonably escape detection? Or, if they do have health concerns, what barriers to care do they experience? Given that all are infertile, why aren’t a greater proportion detected with infertility? Are men with KS less likely to establish relationships as a consequence of health and psychosocial disadvantage? Or are physical examinations and karyotyping not being performed in all assisted reproductive technology programs?
If all cases of KS were identified, what educational, biomedical and psychosocial interventions might be offered, and when? What is the evidence base for each of these?
In an era of rapidly advancing genetic technology and greater understanding of the genetic contribution to disease, opportunities for diagnosis of KS should exist. Given the availability of treatments and interventions for KS — and the possible medical and psychosocial benefits of early diagnosis — avenues for increasing awareness, educating health professionals, and providing appropriate resources for individuals and their families must be developed further. In addition, both the risks and benefits of diagnosis through postnatal, population-based genetic screening for KS should be considered.25
1 Possible phenotypic features of Klinefelter syndrome (KS) according to life stage, and their estimated frequencies*
 |
|
* Features and frequencies that may prompt further clinical investigation by karyotyping. The classic KS phenotype of tall stature, gynaecomastia and undervirilisation is not always apparent. The most universal and reliable indicator of KS is small testicular size. (Adapted from Table 2, Bojesen et al 2007.2) |
2 Distribution of karyotypes in prenatal (1986–2006) and postnatal (1991–2006) diagnoses of Klinefelter syndrome in Victoria, Australia
* Includes karyotypes: 47,XXY/49,XXXXY; |
|||||||||||||||
3 Annual prenatal diagnoses of Klinefelter syndrome (KS), and corresponding prevalences, 1986–2006 in Victoria, Australia
4 Maternal age standardisation of raw prenatal prevalences to estimate the overall age-adjusted prevalence of Klinefelter syndrome (KS) (per 100 000 male births) in Victoria, Australia, 1986–2006
PNT = prenatal testing. * Age-adjusted prevalence rate calculated from these values: 223 per 100 000 male births (1 in 448; 95% CI, 195–254). |
|||||||||||||||
Competing interests
None identified.
Acknowledgements
The authors would like to thank Merilyn Riley of the VBDR, Alice Jaques and Evelyne Muggli of Public Health Genetics at the Murdoch Childrens Research Institute, and staff at the cytogenetics laboratories across Victoria for help with data collection. We are also grateful for the thoughtful suggestions of our reviewers. All authors are supported by funding from the National Health and Medical Research Council of Australia.
References
- Jacobs PA, Strong JA. A case of human intersexuality having a possible XXY sex-determining mechanism. Nature 1959; 183: 302-303. 0_i1096625
- Bojesen A, Gravholt CH. Klinefelter syndrome in clinical practice. Nat Clin Pract Urol 2007; 4: 192-204. 0_i1096627
- Nielsen J, Wohlert M. Sex chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Birth Defects Orig Artic Ser 1990; 26: 209-223. 0_i1096629
- Ratcliffe S. Development of children with sex chromosome abnormalities. Proc R Soc Med 1976; 69: 3. 0_pgfId-1096631
- Hamerton JL, Canning N, Ray M, Smith S. A cytogenetic survey of 14,069 newborn infants. I. Incidence of chromosome abnormalities. Clin Genet 1975; 8: 223-243. 0_pgfId-1096632
- Bochkov NP, Kuleshov NP, Chebotarev AN, et al. Population cytogenetic investigation of newborns in Moscow. Humangenetik 1974; 22: 139-152. 0_pgfId-1106748
- Higurashi M, Iijima K, Ishikawa N, et al. Incidence of major chromosome aberrations in 12,319 newborn infants in Tokyo. Hum Genet 1979; 46: 163-172. 0_pgfId-1106755
- Leonard MF, Schowalter JE, Landy G, et al. Chromosomal abnormalities in the New Haven newborn study: a prospective study of development of children with sex chromosome anomalies. Birth Defects Orig Artic Ser 1979; 15: 115-159. 0_pgfId-1106762
- Maclean N, Harnden DG, Brown WM, et al. Sex-chromosome abnormalities in newborn babies. Lancet 1964; 1: 286-290. 0_i1096636
- Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab 2003; 88: 622-626. 0_i1096638
- Abramsky L, Chapple J. 47,XXY (Klinefelter syndrome) and 47,XYY: estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling. Prenat Diagn 1997; 17: 363-368. 0_i1096640
- Simpson JL, De La Cruz F, Swerdloff RS, et al. Klinefelter syndrome: expanding the phenotype and identifying new research directions. Genet Med 2003; 5: 460-468. 0_i1096642
- Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet 2004; 364: 273-283. 0_i1096644
- Morris JK, Alberman E, Scott C, Jacobs P. Is the prevalence of Klinefelter syndrome increasing? Eur J Hum Genet 2008; 16: 163-170. 0_i1096646
- Ferguson-Smith MA, Yates JR. Maternal age specific rates for chromosome aberrations and factors influencing them. Report of a collaborative European study on 52,965 amniocenteses. Prenat Diagn 1984; 4: 5-44. 0_i1096648
- De Souza E, Morris JK; EUROCAT working group. Case-control analysis of paternal age and trisomic anomalies. Arch Dis Child 2010; Jun 28. 0_i1096650
- Australian Bureau of Statistics. Births Australia 2006. Canberra: ABS, 2006: (ABS Cat. No. 3301.0.) 0_i1096652
- Davey M-A, Taylor O, Oats JJN, et al. Births in Victoria 2005 and 2006. Melbourne: Victorian Perinatal Data Collection Unit, Department of Human Services, 2008. 0_i1096654
- Australian Bureau of Statistics. Year book Australia, 2009–10. (ABS Cat. No. 1301.0.) http://www.abs.gov.au/AUSSTATS/abs@.nsf/allprimarymainfeatures/796378C9B98D7F1CCA 25773700177E5E?opendocument (accessed Nov 2009).
- Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet 2009; 85: 503-514. 0_i1096658
- Denmark S. [Population statistics, 2009] [Danish]. http://www.statistikbanken.dk (accessed Nov 2009).
- Wang YA, Dean JH, Badgery-Parker T, Sullivan EA. Assisted reproduction technology in Australia and New Zealand 2006. Sydney: Australian Institute of Health and Welfare, National Perinatal Statistics Unit; 2006. (AIHW Cat. No. PER 43.) 0_i1096662
- Nielsen J, Pelsen B, Sorensen K. Follow-up of 30 Klinefelter males treated with testosterone. Clin Genet 1988; 33: 262-269. 0_i1096664
- Bojesen A, Juul S, Birkebaek NH, Gravholt CH. Morbidity in Klinefelter syndrome: a Danish register study based on hospital discharge diagnoses. J Clin Endocr Metab 2006; 91: 1254-1260. 0_i1096666
- Herlihy AS, Halliday J, McLachlan RI, et al. Assessing the risks and benefits of diagnosing genetic conditions with variable phenotypes through population screening: Klinefelter syndrome as an example. J Community Genet 2010; 1: 41-46. 0_i1096670
When ‘Liver Enzymes’ Are Not Hepatic: Late-Onset Pompe Disease
Shauna Madigan, Georgina England, Wayne Rankin
Primary Hyperparathyroidism in Adults: Recent Developments in Diagnosis and Management
Elizabeth Wootton, Sunita M. C. De Sousa, Richard L. Prince, Donald S. A. McLeod, David A. Pattison, Mathis Grossmann
Severe Hypoglycaemia Secondary to Chronic Opioid-Induced Hypothalamic–Pituitary–Adrenal Axis Suppression: An Under-Recognised Phenomenon
Michael Do, Annabelle Hayes, Malgorzata Brzozowska
Rethinking diabetes care for Indigenous Australians: the need for Indigenous‐codesigned and led diabetes models of care
Natalie Nanayakkara, Sharon Atkinson‐Briggs, Alicia J Jenkins, Neale D Cohen
The first Australian evidence‐based guidelines on male infertility
Darren J Katz, Liza O’Donnell, Robert I McLachlan, Tim J Moss, Clare V Boothroyd, Veena Jayadev, Sarah R Catford