




A 49-year-old man presented with verapamil toxicity complicated by hypotension and a junctional rhythm, in the context of deliberate self-poisoning with multiple drugs. The patient’s hypotension normalised following the early use of high-dose insulin euglycaemic therapy (HIET), without the need for additional vasopressors; it recurred when HIET was prematurely stopped, and again stabilised when HIET was recommenced. Consideration should be given to the early use of HIET in treating severe calcium channel blocker toxicity, rather than as a last resort after other therapies have failed. (MJA 2009; 191: 350-352)
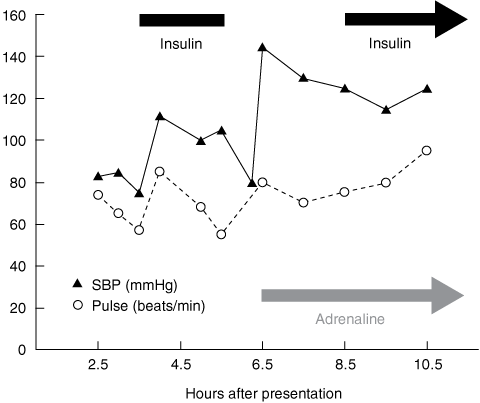
The patient remained hypotensive (BP, 75/45 mmHg; pulse, 56 beats/min) after intubation, so high-dose insulin euglycaemic therapy (HIET) was commenced at 3.5 hours after presentation. He was given dextrose (50 mL 50% glucose) and a 30 IU short-acting insulin IV bolus (~ 0.5 IU/kg), followed by a further bolus of 50 mL 50% glucose and a short-acting insulin IV infusion (30 IU/h) (Box 1). His BP improved to 110/70 mmHg at 4 hours, with a pulse of 82 beats/min and sinus rhythm on ECG, and he remained stable during transfer to the intensive care unit (ICU).
The insulin infusion was abruptly stopped 5.5 hours after presentation, on arrival in the ICU. The patient’s hypotension subsequently recurred (systolic BP, 70 mmHg; pulse, 75 beats/min), prompting administration of 500 mL IV Gelofusine (a colloidal plasma volume substitute; B. Braun, Sydney, NSW) and commencement of an adrenaline IV infusion (20 μg/min). The insulin infusion (30 IU/h) was restarted at 8.5 hours, and his BP again stabilised (Box 1). The propofol IV infusion was gradually increased from 50 mg/h to 150 mg/h between 5.5 hours and 11.5 hours after presentation, and a noradrenaline IV infusion was commenced at 9.5 hours to maintain normotension. At 15.5 hours, pulmonary artery catheter measurements showed a high cardiac index (5.1 L/min/m2; reference range [RR], 2.5–4.0 L/min/m2) and a low systemic vascular resistance index (1047 dynes·s/cm5/m2; RR, 1900–2400 dynes·s/cm5/m2); the patient’s pulse was 85 beats/min and BP was 140/60 mmHg.
HIET is an increasingly accepted therapy for calcium channel blocker (CCB) toxicity, but reports of its use are limited and it remains controversial. Indeed, the scarcity of severe CCB poisoning cases means that a randomised controlled trial of HIET may not be feasible.1 Treating clinicians who seek advice from clinical toxicologists are often hesitant about the high doses required and the potential for adverse effects. Such hesitancy is potentially harmful, as a hypotensive patient with a CCB overdose who otherwise appears well is at risk of abrupt lethal cardiovascular collapse.1 HIET is traditionally recommended after other therapies have failed.2,3 This case report aims to raise awareness of HIET for the treatment of CCB toxicity and supports its early use, rather than as a last resort.4
Verapamil binds the alpha-1 subunit of L-type calcium channels, preventing the intracellular influx of calcium.5 These channels are functionally important in cardiac myocytes, vascular smooth muscle cells, and islet beta cells.5 Verapamil’s cardiac toxicity results from excessive negative inotropy, negative chronotropy and negative dromotropy, characterised by myocardial depression, sinus bradycardia, and atrioventricular node blockade.4 Vascular smooth muscle tone is impaired, resulting in decreased afterload, systemic hypotension, and coronary vasodilation.5
Less well known are the metabolic effects of CCBs such as verapamil. Under the stress of the drug-induced shock state, the cardiac myocytes shift from using free fatty acids, their favoured “resting state” energy substrate, to carbohydrates.3,4 CCB toxicity also impairs the uptake of glucose and free fatty acids by cardiac myocytes3,4 and inhibits calcium-dependent mitochondrial activity required for glucose catabolism.3,4 Furthermore, insulin release is dependent on calcium influx into islet beta cells through L-type calcium channels.3,4 Thus, CCB toxicity can cause hypoinsulinaemia,3,4 which, in conjunction with CCB-induced insulin resistance, may lead to hyperglycaemia and a ketoacidotic state.6
Atropine, calcium boluses and infusions, glucagon, inotropes, vasopressors, and cardiac pacing have all been advocated for managing CCB toxicity, despite questionable efficacy.3,4,7-9 For instance, the evidence for glucagon is limited to small, non-blinded animal studies where no survival benefit or improvement in mean arterial pressure was shown, although heart rate improved in some cases.7 Rarely, heroic measures such as extracorporeal circulatory support and intra-aortic balloon counterpulsation have been successfully employed.5,10
HIET was first used to treat verapamil toxicity in humans in 1993, with a favourable outcome.6 Since then, in addition to animal studies, there have been nearly 70 cases reporting the beneficial use of HIET in humans, with an overall survival rate of 85%.8 However, to our knowledge, use of HIET in humans before the administration of glucagon or vasopressors has only been reported once.6 There have been some reports of HIET failure in treating CCB toxicity, although the dosing of insulin was low or uncertain, or it was used late.6,8 Early use of HIET may be more effective than HIET rescue therapy, as CCB-induced insulin resistance is greatest in the first 24 hours2 and the maximal haemodynamic benefit of HIET may not occur immediately.6
HIET may allow the heart to overcome metabolic starvation in CCB toxicity, which compounds the direct CCB impairment of myocardial contractility.3,4 Insulin increases glucose and lactate uptake by myocardial cells and improves function without increased oxygen demand.11,12 It also induces pyruvate dehydrogenase, hastening myocardial lactate oxidation, and helps clear the cytosol of glycolytic byproducts that impair calcium handling and cause diastolic dysfunction.3 Insulin promotes excitation–contraction coupling and contractility because enhanced glycolysis promotes increased sarcoplasmic reticulum-associated calcium ATPase activity and increased cytoplasmic calcium concentrations, and promotes calcium entrance into mitochondria and sarcolemma.3
HIET may be best used adjunctively with other measures such as catecholamines, for two reasons. First, insulin-mediated inotropy is not catecholamine-mediated, and is not affected by β blockers.3 Second, although insulin appears to improve myocardial contractility, it has no chronotropic effect and may cause vasodilation.3,8
HIET is safe, and adverse events are predictable, uncommon, and easily managed.2,8 The maximum safe dose of insulin is unknown, but loading doses of 0.5–1.0 IU/kg followed by infusions of 0.1–2.5 IU/kg/h are typically used.8 Interestingly, neither the inadvertent administration of a 1000 IU insulin loading dose for verapamil toxicity13 nor treatment of toxic cardiogenic shock for 2 days with a 6 IU/kg/h insulin infusion had any adverse effects.14 Adverse effects of HIET include hypoglycaemia, hypokalaemia, hypomagnesaemia, and hypophosphataemia.2,6,8 Although these are rarely clinically significant, they necessitate careful monitoring. Hypoglycaemia (blood glucose < 3.3 mmol/L) occurred in 16% of 55 published cases,8 and no cases of hypoglycaemia within 24 hours of CCB overdose were noted in Greene and colleagues’ series of seven cases.2 Greene et al also reported a mean dextrose requirement of 0.05 g/kg/h (range, 0–0.17 g/kg/h), although the mean blood glucose level exceeded the euglycaemic range.2 Some cases of severe CCB toxicity in patients presenting with hyperglycaemia do not require any additional glucose administration despite high-dose insulin therapy,15 and hypoglycaemia may be more likely in milder cases without marked hypotension.8 In addition, hypokalaemia (potassium < 3.5 mmol/L) was noted in only two patients in Greene et al’s small series, with a minimum potassium level of 2.8 mmol/L.2 Excessive correction of hypokalaemia should be avoided, because it reflects the intracellular shift of potassium from the extracellular compartment due to the action of insulin, rather than a potassium-depleted state.4 Interestingly, hypokalaemia in HIET may augment myocardial contractility by enhancing calcium entry during systole, and increased intracellular potassium may have a membrane-stabilising effect in excitable cells.4,6
In conclusion, we advocate consideration of the early use of HIET (as detailed in Box 2) for the prevention and treatment of life-threatening complications from potentially lethal CCB overdoses. HIET is safe, inexpensive and freely available, and suitable for use even in remote settings before transfer to a referral centre.
1 Early changes in the patient’s systolic blood pressure (SBP) and heart rate, relative to treatment with high-dose insulin and adrenaline infusions
 | |||||||||||||||
2 Recommended high-dose insulin euglycaemic therapy protocol,3,4,9 based on the clinical experience of the Western Australian Toxicology Service, published case reports, reviews and animal studies
Glucose 25 g (50 mL of 50% solution) IV bolus, unless marked hyperglycaemia (blood glucose > 22 mmol/L) is present
Short-acting insulin 1 IU/kg bolus to maximally saturate insulin receptors
Short-acting insulin infusion starting at 0.5 IU/kg/h and titrated every 30 min to a maximum of 5 IU/kg/h*
Dextrose 25 g/h IV infusion titrated to maintain euglycaemia (blood glucose, 5.5–14 mmol/L); central venous access may be required to allow use of concentrated solutions (eg, 50% dextrose) and limit excess volume administration
Glucose — every 20 min for first hour, then every 1 h
Potassium — replace only if < 2.5 mmol/L and there is a source of potassium loss
Improvement in myocardial ejection fraction (> 50%); increased BP (systolic BP > 90 mmHg in adults)
Adequate heart rate (> 60 beats/min)
Resolution of acidaemia; euglycaemia; adequate urine output (1–2 mL/kg/h)
Reversal of cardiac conduction abnormalities (QRS interval < 120 ms)
Improved mentation
IV = intravenous. BP = blood pressure. * The maximum safe and effective rate of infusion is unknown but may be even higher than 5 IU/kg/h. In animal studies, insulin infusions as high as 10 IU/kg/h have been safely used.11
- 1. Levine MD, Boyer E. Hyperinsulinemia-euglycemia therapy: a useful tool in treating calcium channel blocker poisoning. Crit Care 2006; 10: 149.
- 2. Greene SL, Gawarammana I, Wood DM, et al. Relative safety of hyperinsulinaemia/euglycaemia therapy in the management of calcium channel blocker overdose: a prospective observational study. Intensive Care Med 2007; 33: 2019-2024.
- 3. Megarbane B, Karyo S, Baud FJ. The role of insulin and glucose (hyperinsulinaemia/euglycaemia) therapy in acute calcium channel antagonist and beta-blocker poisoning. Toxicol Rev 2004; 23: 215-222.
- 4. Lheureux PE, Zahir S, Gris M, et al. Bench-to-bedside review: hyperinsulinaemia/euglycaemia therapy in the management of overdose of calcium-channel blockers. Crit Care 2006; 10: 212.
- 5. Salhanick SD, Shannon MW. Management of calcium channel antagonist overdose. Drug Saf 2003; 26: 65-79.
- 6. Yuan TH, Kerns WP 2nd, Tomaszewski CA, et al. Insulin-glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol 1999; 37: 463-474.
- 7. Bailey B. Glucagon in beta-blocker and calcium channel blocker overdoses: a systematic review. J Toxicol Clin Toxicol 2003; 41: 595-602.
- 8. Kerns W 2nd. Management of beta-adrenergic blocker and calcium channel antagonist toxicity. Emerg Med Clin North Am 2007; 25: 309-331; abstract viii.
- 9. Murray L, Daly F, Little M, Cadogan M. Toxicology handbook. Sydney: Elsevier Australia, 2007.
- 10. Baud FJ, Megarbane B, Deye N, Leprince P. Clinical review: aggressive management and extracorporeal support for drug-induced cardiotoxicity. Crit Care 2007; 11: 207.
- 11. Holger JS, Engebretsen KM, Fritzlar SJ, et al. Insulin versus vasopressin and epinephrine to treat beta-blocker toxicity. Clin Toxicol (Phila) 2007; 45: 396-401.
- 12. Kline JA, Leonova E, Raymond RM. Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med 1995; 23: 1251-1263.
- 13. Place R, Carlson A, Leiken J, et al. Hyperinsulin therapy in the treatment of verapamil overdose [abstract]. J Toxicol Clin Toxicol 2000; 38: 576-577.
- 14. Holger JS, Engebretsen KM, Marini JJ. High dose insulin in toxic cardiogenic shock. Clin Toxicol (Phila) 2009; 47: 303-307.
- 15. Boyer EW, Shannon M. Treatment of calcium-channel-blocker intoxication with insulin infusion. N Engl J Med 2001; 344: 1721-1722.
None identified.