





Volume 191 - Issue 1
Evaluation of non-invasive prenatal RHD genotyping of the fetus
Authors: Catherine A Hyland, Glenn J Gardener, Helen Davies, Minna Ahvenainen, Robert L Flower, Darryl Irwin, Jonathan M Morris, Christopher M Ward and Jonathan A Hyett
Med J Aust 2009; 191 (1): 21-25. || doi: 10.5694/j.1326-5377.2009.tb02668.x
Published online: 6 July 2009
Published online: 6 July 2009
Abstract
Objective: To evaluate a non-invasive molecular test using free circulating fetal DNA in maternal plasma to predict the fetal RHD type.
Design: A prospective cohort study.
Participants and setting: Venous blood samples were collected from 140 Rhesus (Rh) D-negative women booked for antenatal care in two tertiary maternity hospitals in Sydney and Brisbane between November 2006 and April 2008. Cell-free DNA, including free maternal and fetal DNA, was extracted from maternal plasma in the tertiary Australian Red Cross Blood Service laboratory, and three exon regions of the RHD gene were amplified.
Main outcome measures: Comparison of the predicted fetal RHD status and the infant’s RhD serotype. Secondary analysis involved using SRY and RASSF1A assays as internal controls to confirm the presence of fetal DNA in RHD-negative samples.
Results: Of 140 samples tested, results for RHD status were assigned for 135, and all 135 predictions were correct. A result was not assigned in five cases: three did not meet strict threshold criteria for classification, and two were due to RHD variants. Fetal SRY status was correctly predicted in 137 of 140 cases. In 16 samples typed both RHD- and SRY-negative, a positive RASSF1A result verified the presence of fetal DNA.
Conclusions: Non-invasive testing of multiple exons provides a robust method of assessing fetal RHD status, and provides a safer alternative to amniocentesis for the management of RhD-negative pregnant women who are isoimmunised.
Despite the introduction of postpartum and antenatal anti-D immunoglobulin prophylaxis for phenotypic Rhesus (Rh) D-negative pregnant women, a small proportion still become sensitised and express antibodies to RhD.1,2 Isoimmunised pregnancies are at high risk of haemolytic disease of the newborn (HDN), which can cause fetal anaemia, hydrops and intrauterine fetal death. Babies born with HDN are at significant risk of neonatal morbidity and mortality. The current strategy for monitoring RhD-negative pregnant women at high risk for HDN relies on serial assessment of maternal antibody levels, Doppler ultrasound measurement of the peak systolic velocity of blood flowing through the fetal middle cerebral artery (which rises in anaemia) and, when necessary, intrauterine fetal blood sampling.3 Antenatal care is more intensive, requiring regular review and investigation in specialist fetal medicine centres, which is inconvenient for patients and may place the pregnancy at risk through invasive testing such as amniocentesis.4,5
RhD-negative pregnant women with a heterozygous partner have a 50% chance of having an RhD-negative fetus that is not at risk of HDN, and these women could be reassured and managed less intensively if this were confirmed. Genotyping to assess fetal RHD status has, until recently, only been available using amniocentesis.6,7 This procedure carries a 1% risk of pregnancy loss and a risk of increasing maternal antibody levels in affected pregnancies.4,5 The discovery in 1997 that cell-free fetal DNA (ffDNA) is present in the maternal circulation has provided a potential method for non-invasive assessment of fetal RHD using a maternal venous blood sample.8,9 Non-invasive fetal RHD genotyping has been successfully developed in specialised laboratories in Europe, where a fetal genotyping service is offered to clinicians. In contrast, real-time polymerase chain reaction (RT-PCR) techniques for RHD genotyping have not been available to date in Australia, although gel electrophoresis has been applied.10-15
Our objective was to evaluate a non-invasive test to assess fetal RHD status in an Australian obstetric population using established RT-PCR protocols, with some system enhancements. These included improvements (compared with international groups) in isolation of ffDNA from maternal plasma, and the use of RT-PCR to amplify three regions of RHD (in comparison with other Australian groups).15 We also added an RASSF1A methylation assessment to the test algorithm to confirm the presence of ffDNA in RHD- and SRY-negative samples, to safeguard against false-negative reporting.16
Methods
Subjects
Venous blood samples were prospectively collected from 140 RhD-negative women presenting for routine obstetric care at two tertiary maternity hospitals in Sydney and Brisbane between November 2006 and April 2008. There were 26 samples taken at 12–16 weeks’ gestation, 61 samples at 17–27 weeks’ gestation, and 53 samples at ≥ 28 weeks’ gestation. Nine women were isoimmunised to RhD.
The study was approved by ethics committees of Mater Health Services, Royal North Shore Hospital and the Australian Red Cross Blood Service (ARCBS). All women gave informed consent.
Sample logistics and plasma preparation
Blood samples (18 mL) were collected in tubes containing EDTA anticoagulant and transported to the tertiary ARCBS laboratory within the timeframes used for screening blood donors for labile viral RNA markers.17 Plasma was separated from the cellular component using a two-step centrifugation protocol and stored at − 70°C. Plasma was frozen within 24 hours of blood sampling, except for three samples that were frozen within 46, 100 and 170 hours.
Review of maternal RhD blood group status
During pregnancy, women who are RhD-negative are identified by routine serological screening. From a clinical perspective, women who have “partial” RhD antigen expression, lacking some but not all of the D epitopes, should be managed according to protocols devised for RhD-negative women. Many hospital laboratories therefore use screening assays that have a lower sensitivity and specificity and do not distinguish between RhD-negative women and those who have an RhD variant. As a common standard, we ensured that all maternal RhD blood group testing was performed according to blood donor screening protocols that are optimised to detect partial and weak D.18 Although not present in this cohort, this included testing for the extreme form of the weak D antigen, DEL, by red cell antibody absorption and elution tests on samples with an RhCe haplotype.
We recognised that the presence of a parental RHD variant may cause errors in fetal genotyping.19-21 To assess the impact of these variants, the study protocol included a procedure for further analysis of samples that gave discrepant or indeterminate results. To allow later comparison with maternal genotype, maternal genomic DNA (mgDNA) was extracted and stored from the white cell “buffy” coat at the time of DNA extraction from maternal plasma.
DNA extraction from maternal plasma
Cell-free DNA, including both free maternal DNA and ffDNA, was extracted from maternal plasma, with each sample extracted in duplicate and the resultant two eluates pooled for same-day fetal RHD genotyping.
Initially, two methods using different QIAamp kits (QIAGEN, Melbourne, Vic) were evaluated with nine samples. The first method, using a QIAamp DNA Blood Mini Kit, isolated DNA from 800 μL of plasma, eluting DNA in a final volume of 55 μL.10 The second, using a QIAamp MinElute Virus Spin Kit, involved a protocol recommended by Sequenom Inc (San Diego, Calif, USA), isolating DNA from 1000 μL plasma and eluting DNA in 55 μL.
Comparison of the two DNA isolation methods showed lower cycle threshold (Ct) values for fetal markers (RHD exons 4, 5 and 10, and SRY) using the QIAamp MinElute Virus Spin Kit (P < 0.001, χ2 goodness-of-fit test). This result is consistent with yielding higher levels of ffDNA, and this method was adopted for the remainder of the study.
Fetal RHD genotyping by RT-PCR
of ffDNA
The presence of fetal RHD sequences was determined with two separate duplex RT-PCR assays, with each test performed in quadruplicate.10,11 One duplex amplified RHD exon 4 and 10 sequences, and the other amplified RHD exon 5 and the Y chromosome-located SRY gene.10,11 RT-PCR was performed using the Rotor-Gene 3000 (Corbett Research, Sydney, NSW), and results were analysed with quantification software.22
Supplemental test algorithm for indeterminate samples
The primer and probe designs for exons 4 and 5 do not permit amplification of the non-functional RHD pseudogene associated with RhD-negative black Africans. Such samples will therefore type as exon 4- and 5-negative and exon 10-positive. In addition, other rearranged RHD-CE-D genes can generate discordance between RT-PCR exon typing and either be non-functional or generate partial D antigens. For example, the gene responsible for the clinically significant RhDVI antigen will also type as exon 4- and 5-negative and exon 10-positive, and be indistinguishable from the pseudogene on the primary RT-PCR test. A follow-up supplemental test algorithm, including RT-PCR using pseudogene primers and probes specific for exons 4 and 5, and also RT-PCR for exon 7, was performed to further resolve such samples. A pseudogene DNA control was provided by Dr Kirstin Finning (International Blood Group Reference Laboratory, Bristol, UK).
Quality control measures
The quality of the primers, probes and RT-PCR reagents was assessed using a defined quantity of purified human male genomic DNA (Promega, Madison, Wis, USA) in a twofold dilution series from 0.02 ng/μL down to 0.001 ng/μL. The RT-PCR duplex assays detected down to 1.5 copies of target genomic RHD and SRY sequence, as reported elsewhere.9 For fetal RHD genotyping, three water (no template), four RhD-negative and three RhD-positive controls were included in each PCR run to monitor for contamination. For each sample, total (maternal and fetal) cell-free DNA yield was quantified by amplifying the chemokine receptor gene (CCR5).10,11 Inclusion of the CCR5 assay helps identify samples with an excess of maternal DNA (eg, from maternal white cell lysis) where there may be interference with detection of ffDNA sequences. In addition, comparison of RHD exon 10 RT-PCR Ct values compared with the CCR5 Ct may signify the presence of a non-functional maternal RHD gene and trigger further mgDNA studies.10,11
RASSF1A test as a control marker for ffDNA
The SRY gene served as an internal control marker to confirm the presence of fetal DNA. To guard against false-negative RHD results, in samples where neither RHD nor SRY sequences were detected, further analysis to show that fetal DNA was present was performed by testing for the presence of a hypermethylated RASSF1A gene. The promoter of the RASSF1A gene, a tumour suppressor gene, is hypermethylated in DNA derived from the placenta and hypomethylated in DNA derived from the mother.16 The test involves digestion of 17.5 μL of plasma-derived DNA with 100 U of BstUI, a methylation-sensitive restriction enzyme, at 60°C for 16 hours. This effectively removes the maternal RASSF1A gene, and the fetal gene can then be detected by RT-PCR. Amplification was performed in tandem with beta-actin, which served as an internal control to test for the completeness of the digestion.16 Tests were performed in triplicate, with each run also including DNA extracted from whole blood derived from an RhD-negative non-pregnant woman as a negative control to further show that DNA digestion was complete.
Interpretation of results of
DNA analysis
Criteria established by Finning and colleagues were used to interpret results.10,11 A fetus was assessed as being RHD-positive if two of the four replicates for each of the 4, 5 and 10 RHD exons plus an additional three replicates from any of the RHD exons gave Ct values < 42. A fetus was assessed as being RHD-negative if at least 11 of 12 RHD replicates gave no Ct values (ie, no amplification) and there was a positive signal for the SRY (≥ 2/4 replicates) or the RASSF1A (≥ 2/3 replicates) genes. Samples not classified as either positive or negative were described as indeterminate, and further testing was performed according to the supplemental algorithm. The accuracy of predicted fetal RHD status was assessed by comparison with the infants’ RhD serotype determined from cord blood after delivery.
Statistical analysis was performed using GraphPad Prism, version 4.03 (GraphPad Software, San Diego, Calif, USA).
Results
Review of maternal RhD status
Extended donor testing of maternal serotype showed one of the 140 apparently RhD-negative mothers to be partial RhD-positive, carrying an RhDVI antigen. This mother, who was white, had an RhD-negative C+c+e+ phenotype. Genotyping confirmed that she was RHD exon 4- and 5-negative and exon 7- and 10-positive, consistent with an RhDVI variant.7,20,21
Accuracy of RHD genotyping
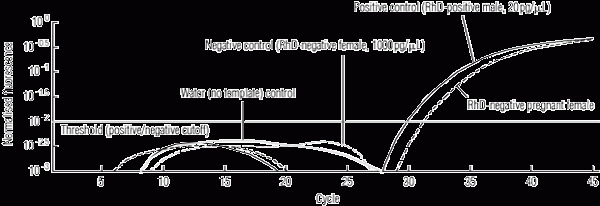
In the first round of analysis, 135 of the 140 RhD-negative women had informative results predicting fetal RHD status and, of these, there was 100% concordance with the infant RhD serotype. Ninety-five fetuses were predicted to be RHD-positive, and an example of the molecular analysis for such a case is shown in Box 1.
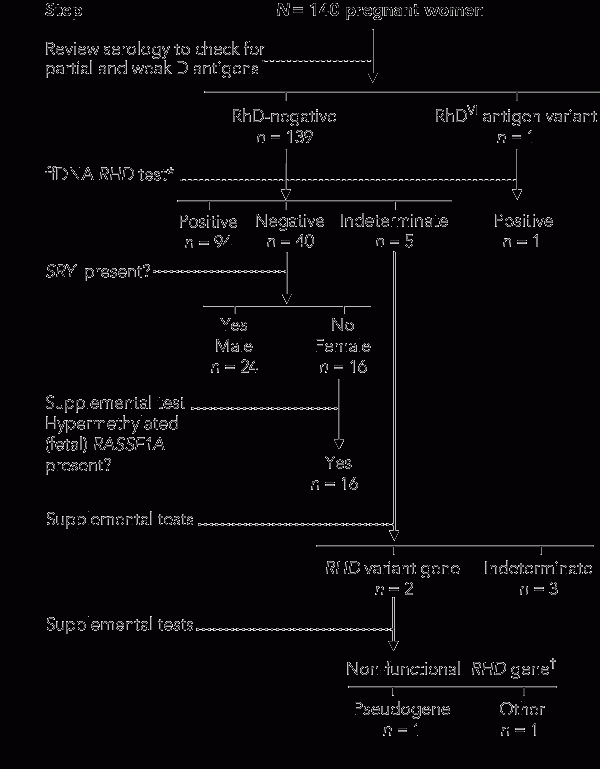
Fetal RHD status could not initially be determined for five of the 140 women. These indeterminate results triggered the supplemental test algorithm, which produced further information about D gene variants in two cases, but failed to resolve the issues of low replicate amplification in the other three. Two of these three samples, collected at 12 and 15 weeks’ gestation, gave positive amplification signals in 7/12 RHD exon replicates, rather than the 9/12 required to state that the fetus was likely to be RHD-positive. Both of these neonates did, however, prove to be RhD-positive in subsequent serotyping. The third indeterminate sample consistently showed low-level signal detection (4/12 across all exons), and the baby’s serotype was found to be RhD-negative.
For the two samples revealing D gene variants, initial testing had shown no amplification of exons 4 and 5, but exon 10 gave a positive signal in all four replicates (Box 2). In one sample, supplemental testing amplified exons 4 and 5 using the pseudogene-specific primers (data not shown). Further assessment of mgDNA showed that the mother was RHD-negative for all three exons. These findings were indicative of paternal inheritance of the pseudogene, consistent with the ethnic origin of the father, and the neonate serotyped RhD-negative as anticipated.
In contrast, supplemental testing of the second variant sample showed no evidence of a pseudogene, and there was no amplification signal for exon 7. Given that the mother serotyped as RhD-negative, and that analysis of mgDNA also showed an exon 4-, 5- and 7-negative but exon 10-positive result, we concluded that this was not a functional RHD gene and the neonate would serotype as RhD-negative, which was confirmed after delivery.
A third RHD variant detected in the study cohort was seen in the mother reclassified as having an RhDVI partial antigen variant. In this case, the fetus had 4/4 positive signals for RHD exons 4, 5 and 10, and 4/4 positive signals for SRY. As exons 4 and 5 were not detected in the mother, we predicted that the fetus was an RHD-positive male, and this was confirmed after birth (Box 2).
Quality assurance
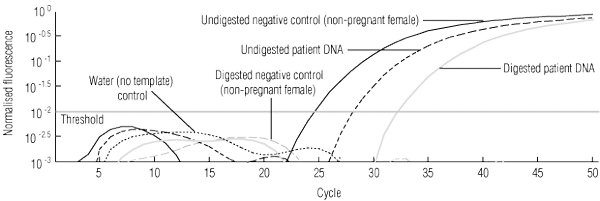
In the 40 women predicted to have an RHD-negative fetus, the presence of fetal DNA was verified by detection of SRY signals for 24, which correlated correctly with delivery of male infants. The presence of fetal DNA for the remaining 16 women with negative SRY signals was verified by detection of the fetal-associated hypermethylated RASSF1A gene (Box 3), and female sex was correctly predicted in all 16 cases.
Using SRY as an internal control to confirm the presence of fetal DNA, post-delivery correlation of predicted fetal sex was correct in 137/140 cases, including all of the 40 predicted RHD-negative cases. The three discrepant cases included one where no assignation could be made for either RHD or SRY. In another case, plasma preparation was delayed until 170 hours after collection and, although an SRY signal was not detected, a male infant was born. Significantly higher total (maternal and fetal) free DNA levels were noted in this sample (2200 ng/mL; mean, 288 ng/mL). The final sample was genotyped as an RHD-positive male, whereas the infant was an RhD-positive female. We were not able to explain this discrepancy.
RASSF1A enzyme digestion and amplification, required as an internal control in the 16 RHD- and SRY-negative samples, was performed on a total of 40 samples (20 RHD-positive and 20 RHD-negative) to further evaluate its robustness. There was good correlation of the Ct values for the RASSF1A and RHD assays for the 20 RHD-positive samples (R2 = 0.9), suggesting both sequences were of fetal origin.16 Dilution studies comparing the sensitivity of the RASSF1A and RHD assays (to determine the risk of a false-negative result) have shown that amplification fails with the RASSF1A probe at the same dilution as the RHD probes, which is important if it is to be relied on as an internal control for non-invasive RHD genotyping.
Discussion
We evaluated an RT-PCR technique for non-invasive prenatal assessment of fetal RhD blood group in an Australian obstetric population, and our strategy of using a predictive algorithm based on analysis of multiple RHD exons allowed fetal RHD status to be predicted in 96% of cases, with all predictions being accurate. Therefore, where RHD status could be predicted, both the positive and negative predictive values were 100% for this sample size. Further expansion of the sample size would increase the power for measuring sensitivity and specificity.
From a clinical perspective, the robustness of such a test is based on risks of false-positive and false-negative reporting. We have shown that RHD variants, which potentially mislead phenotypic representation, can be identified safely through a supplemental algorithm in cases where the initial screening strategy gives an indeterminate result. The potential for a false-negative result arises if there is no ffDNA in the maternal plasma, and the addition of the RASSF1A assay to serve as a universal ffDNA control improved our ability to exclude this situation. In clinical practice, a universal marker of fetal DNA such as RASSF1A is important in excluding a false-negative result in RHD-negative females.
A false-negative result could also occur if low levels of ffDNA were not amplified. Preliminary studies using this assay established that the RT-PCR method detected down to 1.5 target copies of genomic DNA. Although we recognise that the assay may be less robust when amplifying fragments of ffDNA, there is currently no universally accepted international standard to determine this. However, ffDNA was detected across the full gestational age range (12–36 weeks) and there was only one false-negative error where SRY failed to amplify, in a sample for which processing was delayed (170 hours) and total free DNA levels were much higher than normal. Even so, RHD was detected correctly for this case, reflecting the test optimisation for RHD (but not SRY). Similar findings have been reported when sample processing is delayed, highlighting the importance of timely processing in clinical applications.23 The sensitivities of the RASSF1A and RHD assays were comparable, thereby negating the risk of false-negative reporting at low fetal DNA concentrations. As others have noted, because the test involves the promoter region of a tumour suppressor gene, it may be of no value in pregnant women who have had cancer, and determining such a medical history may be important.16,24
We aimed to correlate the predicted fetal RHD genotype with the RhD serotype determined from cord blood. It is important to appreciate that there are limitations in establishing RhD serotype using standard qualitative methods that vary in their sensitivity. Indeed, we would advocate that ffDNA analysis should be performed in conjunction with extended maternal serotyping to check for partial D, weak D and DEL variants. This was reflected in the reclassification of one mother as a DVI variant. This could have consequences for clinical management in the event of fetal–maternal haemorrhage, as such women may need larger doses of anti-D to neutralise the fetal RhD-positive cells. Altogether, three samples (2%) had evidence of RHD variants within the family, and in two of these the primary test gave an indeterminate result for fetal RHD status. The supplemental screening process did, however, enable us to determine both maternal and fetal genotype in all cases.
It has been suggested that ffDNA analysis of fetal RHD status could be applied in population-based screening programs to limit the amount of prophylactic anti-D needed to prevent isoimmunisation during pregnancy.23 The prevalence of RhD variants in the Australian population is incompletely defined, but our study suggests that the number of cases needing intensive follow-up through the supplementary algorithm would not be insignificant.
This assay has primarily been designed to accurately report fetal RHD genotype, but it may also be useful for the determination of fetal sex. Accurate sex determination would be valuable to couples at risk of having a child with an X-linked disease, and if this information was available at or before 12 weeks’ gestation, it could potentially halve the number of invasive chorionic villus sampling tests performed on this basis.25 Although the assay accurately identified sex in 98% of cases, we would not yet recommend its introduction for this purpose, and we plan to investigate both the addition of other male-specific genes and the sensitivity of the test at < 12 weeks’ gestation.
The inclusion in our study of assays for three RHD exons covers population variants, and the use of SRY and RASSF1A control genes provides security against false-negative results. Within the study parameters, the results are applicable for sample collection in the gestational age range of 12–36 weeks and with plasma preparation within 24 hours of sample collection. The accurate determination of fetal sex needs further assessment, particularly at earlier gestational ages and ideally with more than one Y chromosome locus being assessed. The effectiveness of the RASSF1A assay may allow its application to other obstetric indications, to assess where quantification of ffDNA levels is valuable. The results of this study warrant the clinical inclusion of this test in the assessment of isoimmunised RhD-negative women known to be at risk of HDN. The benefits of non-invasive fetal RHD genotyping include reduced fetal surveillance for those who are assessed as RHD-negative and, in particular, will avoid the potential dangers of amniocentesis.
1 Detection of fetal-derived RHD exon 5 sequences in an RhD-negative pregnant woman, by fluorescence versus cycle number and showing assay controls
2 Decision tree for fetal RHD genotyping in RhD-negative pregnant women
3 Hypermethylated RASSF1A test system to confirm presence of free fetal DNA (ffDNA) in samples testing as RHD- and SRY-negative*
 | |||||||||||||||
|
* Multiplex real-time polymerase chain reaction for the RASSF1A promoter gene using FAM- and VIC-labelled probes and using DNA both undigested and digested with BstUI.16 The beta-actin gene was used as an internal control to show that enzyme digestion was complete (patterns not shown). The analysed data show the signals crossing the threshold for these samples: undigested negative-control DNA (5 ng) from a non-pregnant RhD-negative female — note that after digestion, the signal does not cross the threshold; undigested patient DNA (0.45 ng) — this signal represents amplification of RASSF1A from plasma-derived ffDNA and maternal DNA; and digested patient DNA (0.47 ng) — the signal above threshold remaining after BstUI digestion shows that ffDNA is present, as the fetal gene is hypermethylated and enzyme-resistant; therefore samples typing RHD- and SRY-negative are predictive of a female RhD-negative fetus. | |||||||||||||||
Competing interests
None identified.
Acknowledgements
This study was supported by an Australian New Zealand Society of Blood Transfusion scholarship. Sincere thanks to Ms Jocelyn Sedgley, Royal North Shore Hospital, and Ms Glenda Millard, ARCBS, for their energy and commitment to this study. We also thank Dr Geoff Daniels, Mr Peter Martin and Dr Kirstin Finning, International Blood Group Reference Laboratory, United Kingdom, for their support in the development of the methodology, and Dr Allen Chan and Professor Dennis Lo for their advice in the development of the RASSF1A assay.
References
- Clarke CA, Whitfield AG, Mollison PL. Deaths from Rh haemolytic disease in England and Wales in 1984 and 1985. BMJ 1987; 294: 1001. 0_CIGJIGCA
- Robson SC, Lee D, Urbaniak S. Anti-D immunoglobulin in RhD prophylaxis. Br J Obstet Gynaecol 1998; 105: 129-134. 0_CIGCACIF
- Schumacher B, Moise KJ Jr. Fetal transfusion for red blood cell alloimmunization in pregnancy. Obstet Gynecol 1996; 88: 137-150. 0_i1092051
- Tabor A, Philip J, Madsen M, et al. Randomised controlled trial of genetic amniocentesis in 4606 low-risk women. Lancet 1986; 1: 1287-1293. 0_i1092053
- Tabor A, Bang J, Nørgaard-Pedersen B. Feto-maternal haemorrhage associated with genetic amniocentesis: results of a randomized trial. Br J Obstet Gynaecol 1987; 94: 528-534. 0_i1092055
- Bennet PR, Le Van Kim C, Colin Y, et al. Prenatal determination of fetal RhD type by DNA amplification. N Engl J Med 1993; 329: 607-610. 0_i1092057
- Chan FY, Cowley NM, Wolter L, et al. Prenatal RHD gene determination and dosage analysis by PCR: clinical evaluation. Prenat Diagn 2001; 21: 321-326. 0_i1092059
- Lo YMD, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet 1997; 350: 485-487. 0_i1092061
- Lo YMD, Hjelm MN, Fidler C, et al. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. N Engl J Med 1998; 339: 1734-1738. 0_i1092063
- Finning K, Martin P, Daniels G. A clinical service in the UK to predict fetal Rh (Rhesus) D blood group using free fetal DNA in maternal plasma. Ann N Y Acad Sci 2004; 1022: 119-123. 0_i1092065
- Finning KM, Martin PG, Soothill PW, Avent ND. Prediction of fetal D status from maternal plasma: introduction of a new noninvasive fetal RHD genotyping service. Transfusion 2002; 42: 1079-1085. 0_i1092067
- Rouillac-Le Sciellour C, Puillandre P, Gillot R, et al. Large-scale pre-diagnosis study of fetal RHD genotyping by PCR on plasma DNA from RhD-negative pregnant women. Mol Diagn 2004; 8: 23-31. 0_pgfId-1098723
- Bianchi DW, Avent ND, Costa JM, van der Schoot CE. Noninvasive prenatal diagnosis of fetal Rhesus D: ready for Prime(r) Time. Obstet Gynecol 2005; 106: 841-844. 0_pgfId-1098733
- Geifman-Holtzman O, Grotegut CA, Gaughan JP. Diagnostic accuracy of noninvasive fetal Rh genotyping from maternal blood — a meta-analysis. Am J Obstet Gynecol 2006; 195: 1163-1173. 0_pgfId-1098740
- Nelson M, Eagle C, Langshaw M, et al. Genotyping fetal DNA by non-invasive means: extraction from maternal plasma. Vox Sang 2001; 80: 112-116. 0_i1092072
- Chan KCA, Ding C, Gerovassili A, et al. Hypermethylated RASSF1A in maternal plasma: a universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clin Chem 2006; 52: 2211-2218. 0_i1092074
- Mison L, Seed CR, Margaritis AR, Hyland C. Nucleic acid technology screening of Australian blood donors for hepatitis C and human immunodeficiency virus-1 RNA: comparison of two high-throughput testing strategies. Vox Sang 2003; 84: 11-19. 0_i1092076
- Scientific Subcommittee of the Australian and New Zealand Society of Blood Transfusion. Guidelines for pretransfusion testing. 4th ed. Sydney: ANZSBT, 2002. 0_i1092078
- Hyland CA, Wolter LC, Saul A. Three unrelated Rh D gene polymorphisms identified among blood donors with Rhesus CCee (r’r’) phenotypes. Blood 1994; 84: 321-324. 0_i1092080
- Wagner FF, Flegel WA. Review: the molecular basis of the Rh blood group phenotypes. Immunohematology 2004; 20: 23-36. 0_i1092082
- Westhoff CM. The Rh blood group system in review: a new face for the next decade. Transfusion 2004; 44: 1663-1673. 0_i1092084
- Reynisson E, Josefsen MH, Krause M, Hoorfar J. Evaluation of probe chemistries and platforms to improve the detection limit of real-time PCR. J Microbiol Methods 2006; 66: 206-216. 0_i1092086
- Finning K, Martin P, Summers J, et al. Effect of high throughput RHD typing of fetal DNA in maternal plasma on use of anti-RhD immunoglobulin in RhD negative pregnant women: prospective feasibility study. BMJ 2008; 336: 816-818. 0_i1092088
- Chan KCA, Lai PBS, Mok TSK, et al. Quantitative analysis of circulating methylated DNA as a biomarker for hepatocellular carcinoma. Clin Chem 2008; 54: 1528-1536. 0_i1092090
- Hyett JA, Gardener G, Stojilkovic-Mikic T, et al. Reduction in diagnostic and therapeutic interventions by non-invasive determination of fetal sex in early pregnancy. Prenat Diagn 2005; 25: 1111-1116. 0_i1092094