




Most randomised trials are superiority trials, which assess whether a new treatment is more efficacious than a current standard treatment or placebo. However, there is increasing interest in determining whether a new treatment — pharmacological or non-pharmacological — is similar to (equivalent) or no worse than (non-inferior) the standard in terms of efficacy, but preferable owing to lower cost, fewer side effects, easier administration or less harm (Box 1).1-5 Equivalence and non-inferiority trials assess whether the effects of a new treatment, compared with a standard treatment as the active control, stay within or go beyond a predefined clinically acceptable margin — the equivalence or non-inferiority margin. These study designs are useful in situations where a placebo or no-treatment group is considered unethical, such as treating patients with myocardial infarction, AIDS, tuberculosis or cancer. Another driver is the mandatory requirement of regulatory and licensing agencies for comparisons of new treatments with existing treatments.6
Superior efficacy of the standard treatment over placebo has been convincingly proven for a given indication in previous trials.
Efficacy of the standard treatment will be preserved under the conditions of the equivalence or non-inferiority trial.
If the new treatment is shown to have equivalent or non-inferior efficacy, then it too would exhibit superior efficacy to placebo if a placebo-controlled trial were to be performed.
These limitations accentuate the risk of bias in trials that are deficient in design, conduct and reporting. Accordingly, the CONSORT guidelines for randomised trials7 have recently been extended to cover equivalence and non-inferiority trials.8 The aim of this review is to highlight the most critical issues that influence validity and generalisability of these trials.
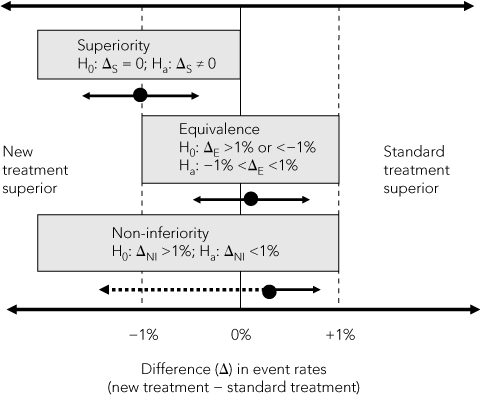
In superiority trials, a minimum clinically important difference between two treatments is hypothesised and, because the new treatment could be either better or worse than the standard treatment, two-sided statistical tests are used to test the null hypothesis (H0) of no difference between treatments (Box 2). This difference is usually measured in absolute units (eg, 2 percentage points for a mortality rate or 5 points on a symptom scale), but can be expressed in relative terms (relative risk or odds ratio). The sample size needed to show a difference, if one exists, is calculated from the hypothesised minimum difference, estimates of event rates in the standard treatment group, numbers of participants who might drop out or cross over between treatments, and the chosen level of statistical significance (usually 5%).
I will not focus on the well established quality criteria for superiority trials7,9 that apply equally to equivalence and non-inferiority trials. Here, I will discuss key elements specific to non-inferiority trials (as these increasingly predominate over equivalence trials), with reference to a case study (Box 3).10-14
Objectives and outcomes. The study protocol should specify that testing for non-inferiority between two active treatments is one objective — or the only objective — and justify the absence of an inactive control group. Measures of the primary (efficacy) outcome (eg, rates of death or specific clinical events) and of secondary outcomes (eg, costs, side effects, patient adherence, safety) should be clearly defined. In some situations, such as the case study in Box 3, greater safety (eg, fewer major bleeds) at the cost of similar or lower efficacy (eg, fewer strokes) can be assessed as a combined endpoint that measures an adjusted or weighted algebraic trade-off of the two items. The extent to which outcome measures and their methods of ascertainment are similar to those used in the original placebo-controlled trials of standard treatment should also be stated.
Clinical judgement: the non-inferiority margin should be the smallest clinically meaningful difference between treatments after considering the seriousness of the primary and secondary outcomes. Margins for mortality or disabling events should be more stringent than those for symptom control or quality of life. For serious efficacy endpoints, many experts stipulate that the margin should be no more than 50%, and preferably no more than 20%, of the treatment effect of the standard treatment, as established in placebo-controlled superiority trials.15 However, no validated rule for calculating the margin currently exists, and many trials use margins that statisticians regard as too liberal.16 Wherever possible, the margin should be validated by published expert consensus,17 and not left to the sole discretion of the investigators and sponsors.
Statistical reasoning: as the magnitude of the standard treatment effect directly influences calculation of the non-inferiority margin, it should be calculated as precisely as possible. Reference should be made to a meta-analysis of all placebo-controlled trials of the standard treatment, in which a summary estimate of effect and its 95% CI are calculated using a random-effects model that demarcates the widest boundaries of uncertainty around the point estimate of effect (Box 4).18 When individual trials have heterogeneous results, the summary estimate should be expressed in both absolute and relative terms. The non-inferiority margin should preferably be no greater than half of the lower limit of the 95% CI of the standard treatment effect.15 Extrapolating this treatment effect from historical superiority trials to a non-inferiority trial involves two assumptions. First, the characteristics of the historical trials closely resemble those of the non-inferiority trial — this is termed “constancy”. Second, both trials are capable of distinguishing between effective and ineffective treatments — “assay sensitivity”. As previously discussed, these assumptions cannot be verified in the absence of a placebo control group.
Blinding. In contrast to unequivocal endpoints such as death, endpoints requiring subjective interpretation are more vulnerable to bias. In a superiority trial, this bias can be minimised by randomising and concealing allocation and blinding outcome assessors, which makes it impossible to know which participants will be, or were, allocated to a particular treatment. However, no such protection exists in a non-inferiority trial. Even with blinding, investigators could potentially randomly discount a significant proportion of endpoints as not meeting pre-specified event definitions, knowing that this will bias the results towards showing non-inferiority. Unblinded trials with highly subjective endpoints are especially susceptible.19 Consequently, quality-control procedures and endpoint assessment must be rigorous and at “arms length” from investigators and sponsors.
In general, the rigour of equivalence and non-inferiority trials is suboptimal. In a review of 88 “equivalence” trials published between 1992 and 1996, 67% inappropriately claimed to be equivalence trials, based on non-significant tests of superiority, and only 22% pre-specified equivalence aim, margin and sample size and actually tested the equivalence hypothesis.20 Eight years later, in a review of 162 trials published during 2003–2004 (46 equivalence; 116 non-inferiority), 93% pre-specified a margin and 78% described sample size calculation.21 However, only 20% of trials justified the choice of margin, only 43% provided both ITT and PP analyses and only 20% fulfilled all key quality criteria discussed above, and of these, 12% stated misleading conclusions.
Many experts express unease about the validity and ethics of equivalence and non-inferiority trials. Criticisms include false pretexts for non-inferiority testing based on commercial rather than patients’ interests, potentially important treatment differences being obscured by liberal non-inferiority margins, unreliable effect estimates based on questionable methods (particularly when standard treatment effects were already small), and betrayal of patient trust by failing to ask and reliably answer important clinical questions.22 The ethics of not using placebo groups in situations where no standard treatment exists or event rates vary widely has also been challenged.23 The lack of sound clinical judgement in choosing margins of difference, disparities between initial study protocols and final analyses, inconsistencies in sample size calculation and use of statistical tests, and failure to include appropriate patient populations or deal with potential confounders have also been highlighted.24,25
1 Contemporary examples of randomised equivalence and non-inferiority trials
2 Comparison of superiority, equivalence and non-inferiority* hypotheses based on a 2% margin of difference in event rates
 |
|
|
3 Case study of two non-inferiority trials
In the treatment of patients with non-valvular atrial fibrillation, the oral direct thrombin inhibitor ximelagatran offers several advantages over warfarin: no need for anticoagulant monitoring, fixed dosing, and less variation in effect with potentially less bleeding risk. Two large clinical trials, one open-label (SPORTIF III) 10 and one double-blind (SPORTIF V) 11 compared the two agents using a non-inferiority design and reported results for the primary efficacy outcome of stroke or systemic thromboembolism (Table). In both studies, the investigators concluded that ximelagatran was as effective as warfarin, but closer inspection of their study design reveals serious deficiencies.
Non-inferiority margin: The margin chosen for both trials was an absolute increase in thromboembolic events of 2% per year. The SPORTIF steering group of 11 members, five of whom were employees of the pharmaceutical sponsor, chose this margin despite citing the results of a meta-analysis of placebo-controlled trials of warfarin in atrial fibrillation that showed, using a fixed-effects model, an absolute reduction in the annual rate of stroke of 3.1%.12 A subsequent random-effects meta-analysis of the same trials13 showed a 2.8% decrease in the annual event rate, with a 95% CI of 1.4%–4.2%. Using the liberal 50% rule, the margin should have been no more than half the lower confidence limit: 1.4% ÷ 2 = 0.7%. This is well below the selected margin of 2%. If a more stringent 1% margin had been chosen, the test for non-inferiority would have failed.
Sample size: For SPORTIF V to show non-inferiority at a margin of 1% with 90% power, it would have required a sample size of more than 7000 participants,14 whereas 3156 were recruited. This low sample size was based on an expected annual event rate for the warfarin group of 3.1%, whereas the observed annual event rate was 1.2%, much closer to the pooled historical annual event rate of 1.9%.
Results from non-inferiority trials comparing ximelagatran with warfarin in patients with non-valvular atrial fibrillation
Primary efficacy outcome event rate* |
|||||||||||||||
4 Steps in defining the non-inferiority margin
A. Meta-analysis of standard treatment effects reported in historical trials 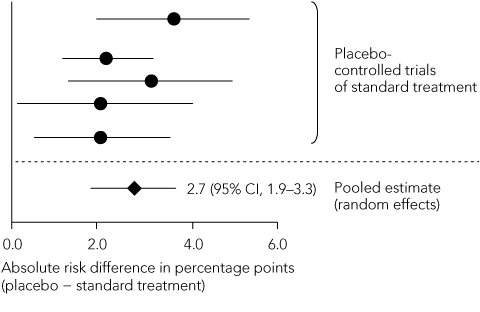 | |||||||||||||||
|
B. Non-inferiority test of new versus standard treatment 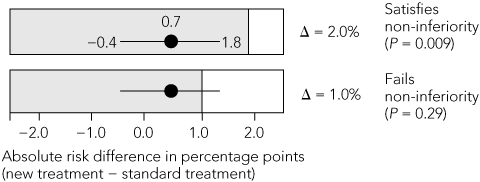 | |||||||||||||||
Adapted with permission from: Kaul S, Diamond GA. Good enough: a primer on the analysis and interpretation of noninferiority trials. Ann Intern Med 2006; 145: 62-69.18 The American College of Physicians is not responsible for the accuracy of this adaptation. | |||||||||||||||
- Ian A Scott1,2
- 1 Department of Internal Medicine and Clinical Epidemiology, Princess Alexandra Hospital, Brisbane, QLD.
- 2 School of Medicine, University of Queensland, Brisbane, QLD.
None identified.
- 1. The SPACE Collaborative Group. 30 day results from the SPACE trial of stent-protected angioplasty versus carotid endarterectomy in symptomatic patients: a randomised non-inferiority result. Lancet 2006; 368: 1239-1247.
- 2. Walsh TJ, Pappas P, Winston DJ, et al for the National Institute of Allergy and Infectious Diseases Mycoses Study Group. Voriconazole compared with liposomal amphotericin B for empirical antifungal therapy in patients with neutropenia and persistent fever. N Engl J Med 2002; 346: 225-234.
- 3. Pfeffer MA, McMurray JJV, Velazquez EJ, et al for the Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349: 1893-1906.
- 4. Righini M, Le Gal G, Aujesky D, et al. Diagnosis of pulmonary embolism by multidetector CT alone or combined with venous ultrasonography of the leg: a randomised non-inferiority trial. Lancet 2008; 371: 1343-1352.
- 5. Kinley H, Czoski-Murray C, George S, et al on behalf of the OpCheck Study Group. Effectiveness of appropriately trained nurses in preoperative assessment: randomised controlled equivalence/non-inferiority trial. BMJ 2002; 325: 1323-1327.
- 6. Pater C. Equivalence and noninferiority trials — are they viable alternatives for registration of new drugs? (III). Curr Control Trials Cardiovasc Med 2004; 5: 8.
- 7. Begg C, Cho M, Eastwood S, et al. Improving the quality of reporting of randomized controlled trials: the CONSORT statement. JAMA 1996; 276: 637-639.
- 8. Piaggio G, Elbourne DR, Altman DG, et al; CONSORT Group. Reporting of noninferiority and equivalence randomized trials: an extension of the CONSORT statement. JAMA 2006; 295: 1152-1160.
- 9. Keech A, Gebski V, Pike R. Interpreting and reporting clinical trials: a guide to the CONSORT statement and the principles of randomised controlled trials. Sydney: MJA Books, 2007.
- 10. Executive Steering Committee on behalf of the SPORTIF III Investigators. Stroke prevention with the oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with non-valvular atrial fibrillation (SPORTIF III): randomised controlled trial. Lancet 2003; 362: 1691-1698.
- 11. Albers GW, Diener HC, Frison L, et al; SPORTIF Executive Steering Committee for the SPORTIF V Investigators. Ximelagatran vs warfarin for stroke prevention in patients with nonvalvular atrial fibrillation: a randomized trial. JAMA 2005; 293: 690-698.
- 12. Halperin JL; Executive Steering Committee, SPORTIF III and V Study Investigators. Ximelagatran compared with warfarin for prevention of thromboembolism in patients with nonvalvular atrial fibrillation: rationale, objectives, and design of a pair of clinical studies and baseline patient characteristics (SPORTIF III and V). Am Heart J 2003; 146: 431-438.
- 13. Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomised controlled trials. Arch Intern Med 1994; 154: 1449-1457.
- 14. Kaul S, Diamond GA, Weintraub WS. Trials and tribulations of non-inferiority: the ximelagatran experience. J Am Coll Cardiol 2005; 46: 1986-1995.
- 15. Committee for Medicinal Products for Human Use. Guideline on the choice of the non-inferiority margin. London: European Medicines Agency, 2005. http://www.emea.europa.eu/pdfs/human/ewp/215899en.pdf (accessed Aug 2008).
- 16. Lange S, Freitag G. Choice of delta: requirements and reality — results of a systematic review. Biom J 2005; 47: 12-27.
- 17. Wyrwich KW, Spertus JA, Kroenke K, et al. Clinically important differences in health status for patients with heart disease: an expert consensus panel report. Am Heart J 2004; 147: 615-622.
- 18. Kaul S, Diamond GA. Good enough: a primer on the analysis and interpretation of noninferiority trials. Ann Intern Med 2006; 145: 62-69.
- 19. Wood L, Egger M, Gluud LL, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ 2008; 336: 601-608.
- 20. Greene WL, Concato J, Feinstein AR. Claims of equivalence in medical research: are they supported by the evidence? Ann Intern Med 2000; 132: 715-722.
- 21. Le Henanff A, Giraudeau B, Baron G, Ravaud P. Quality of reporting of noninferiority and equivalence randomized trials. JAMA 2006; 295: 1147-1151.
- 22. Garattini S, Bertele V. Non-inferiority trials are unethical because they disregard patients’ interests. Lancet 2007; 370: 1875-1877.
- 23. Tramèr MR, Reynolds DJ, Moore RA, McQuay HJ. When placebo controlled trials are essential and equivalence trials are inadequate. BMJ 1998; 317: 875-880.
- 24. Gotzsche PC. Lessons from and cautions about noninferiority and equivalence randomised trials. JAMA 2006; 295: 1172-1174.
- 25. Garrett AD. Therapeutic equivalence: fallacies and falsification. Stat Med 2003; 22: 741-762.
Abstract
New treatments that are potentially as effective as existing treatments are increasingly being developed, some of which may be preferred because of lower cost, fewer side effects, easier administration or less harm.
Non-inferiority trials attempt to establish whether or not a new treatment — drug or non-drug — is no worse than an established treatment for which efficacy has been determined in placebo-controlled trials.
Critical issues in the design and conduct of non-inferiority trials include:
defining the acceptable margin of adverse events that, if exceeded, will render the new treatment inferior to the standard treatment (the non-inferiority margin);
calculating the sample size needed to demonstrate non-inferiority;
assessing the robustness of results in terms of absolute versus relative effects, intention-to-treat versus per-protocol analyses, one-sided versus two-sided statistical tests, and observed versus expected event rates for standard treatment;
evaluating all relevant outcomes, including harm; and
stating conclusions that are consistent with aims and results.
Many non-inferiority trials fail to meet basic quality criteria, report biased and misleading conclusions, and are unduly influenced by commercial sponsors, with some commentators going so far as labelling them unethical.
Clinicians and trial investigators need to exercise caution when interpreting results of non-inferiority trials which, because they lack a placebo group, can only provide an indirect assessment of the efficacy of a new treatment compared with an existing standard, and where the choice of non-inferiority margin can be highly subjective.